Induced pluripotent stem cells (iPSCs) and modeling neurodegenerative disease in the dish
iPSCs and their potential for resetting the biological clock.
Introduction
Due to the limited access to neuronal samples from postmortem organs, understanding of the cellular and molecular mechanisms of the central nervous system (CNS) is difficult.
A significant contribution to elucidating the pathogenesis of various neurological abnormalities has been achieved by transgenic animal models that can mimic human diseases. However, animal models do not fully Replicate complex human disease phenotypes and are very often limited to monogenic disorders. Recent discoveries in pluripotent stem cell technology provide a new opportunity to overcome these limitations. The production of human pluripotent stem cells (hPSCs) from different somatic lineages is a novel tool that may help in the discovery of new treatments for human neurological disorders through disease modeling, drug screening, and regenerative medicine (Figure 1).
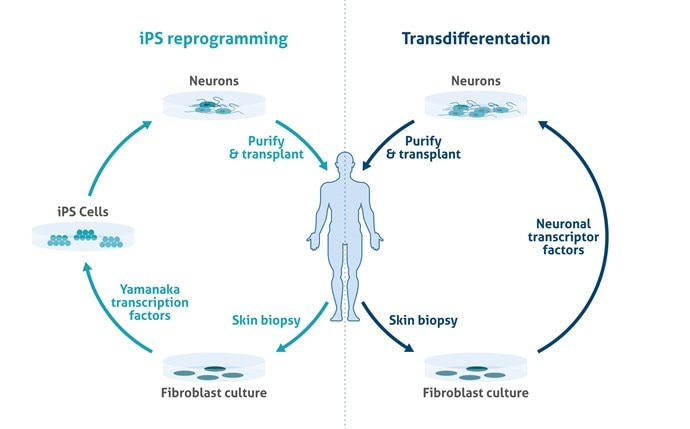
Figure 1. Schematic overview of potentials of induced pluripotent stem cells (iPSC) in relation to neurodegenerative disorders.
We have a broad number of mature neuronal subtypes (synaptophysin Figure 2; Neurofilament medium (NF-M) Figure 3), dopaminergic, cholinergic (CHAT Figure 4), glutamatergic, GABAergic (GAD65 Figure 5; VGAT Figure 6), motor neurons, peripheral sensory neurons, and medium spiny neurons of the striatum. Each of the subtype displays characteristic markers pattern (Figure 7).
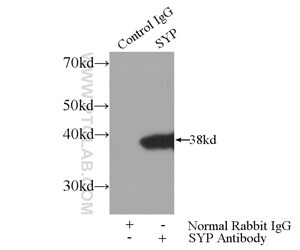
Figure 2. IP Result of anti-Synaptophysin; SYP (IP:17785-1-AP, 3ug; Detection:17785-1-AP 1:1000) with mouse brain tissue lysate 4000ug.
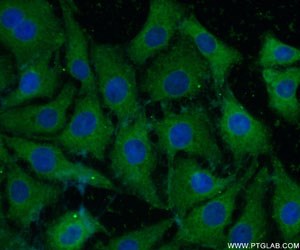
Figure 3. Immunofluorescent analysis of ( -20℃ Ethanol ) fixed SH-SY5Y cells using 20664-1-AP(NF-M-Specific antibody) at a dilution of 1:50 and Alexa Fluor 488-conjugated AffiniPure Goat Anti-Rabbit IgG(H+L).

Figure 4. Rat brain tissue was subjected to SDS PAGE followed by western blot with 20747-1-AP( CHAT Antibody) at a dilution of 1:600 incubated at room temperature for 1.5 hours.
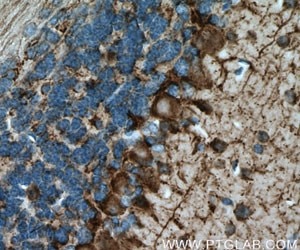
Figure 5. Immunohistochemistry of paraffin-embedded mouse brain tissue slide using 21760-1-AP (GAD65 antibody) at a dilution of 1:200 (under 40x lens) heat mediated antigen retrieved with Tris-EDTA buffer(pH9).
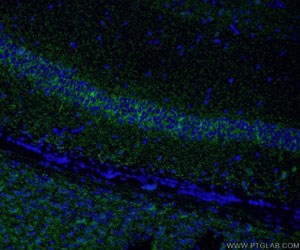
Figure 6. Immunofluorescent analysis of (4% PFA) fixed rat brain tissue using 14471-1-AP (VGAT Antibody) at a dilution of 1:50 and Alexa Fluor 488-conjugated AffiniPure Goat Anti-Rabbit IgG(H+L).
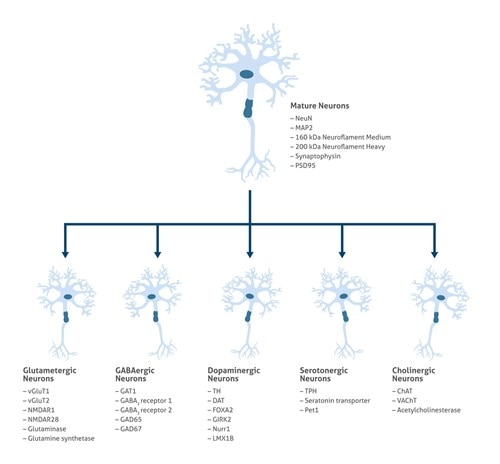
Figure 7. Neuronal subtypes relevant for neurologic diseases modeling.
Alzheimer’s disease
Alzheimer’s disease (AD) is the most common age-associated disorder and is characterized by loss of memory and communication abilities and a change of personality (1). Aging, environmental factors, epigenetic factors, and mutations in the polymorphic apolipoprotein E (APOE) gene are thought to be risk factors for sporadic AD (2). Mutations of the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) are the most frequent reasons for familial AD. Yagi et al. established the first proof-of-principle of iPSC-based modeling when they generated iPSCs from familial Alzheimer’s disease (fAD) patients carrying mutations in the AD causative genes PSEN1 (A264E) and PSEN2 (N141I). The supernatant of neuronal cultures from the AD lines exhibited an elevated ratio of extracellular Aβ42 to Aβ40 compared to controls (3), which was observed in AD patients. Conversely, the treatment of a γ-secretase inhibitor significantly lowered the Aβ levels and the Aβ42/Aβ40 ratio. Sproul et al. developed additional patient-iPSC lines with mutations in PSEN1 (A264E and M146L) and found similar Aβ42/Aβ40 ratios in the neural progenitor cells derived from AD iPSCs compared to the controls (4). In 2012, Israel et al. generated iPSCs from two fAD patients carrying APP duplication (APPDp) and two sporadic Alzheimer’s disease (sAD) patients (5). Choi et al. employed a three-dimensional (3D) culture system for neural induction and differentiation monitoring of the disease development in vitro, which is thought to closely resemble an in vivo environment. By using the 3D culture system, Choi et al. were able to observe amyloid-β plaques and filamentous tau, which are the two best-known hallmarks of AD.
Parkinson’s Disease (PD)
PD is the second most common age-associated progressive neurodegenerative disorder affecting the elderly population. The pathology of PD involves mitochondrial dysfunction, accumulation of α-synuclein misfolded protein (Lewy bodies), and tau phosphorylation, which finally results in the death of dopaminergic neurons located in the substantia nigra of the midbrain (6). Recently, Devine et al. used iPSC-derived neurons as a model to mimic the progress of PD in vitro. Their results demonstrated that a higher expression of α-synuclein could be detected in PD patient-derived midbrain dopaminergic neurons when compared to such neurons derived from an unaffected relative (7).
Amyotrophic lateral sclerosis (ALS)
ALS is an adult-onset, rapidly progressing, fatal neurodegenerative disease. The symptoms are characterized by muscle wasting, which results in difficulty in speaking, swallowing, and, eventually, breathing. Genome-wide association studies (GWAS) have identified several candidate genes as potentially underlying ALS: Superoxide dismutase1 (SOD1), TAR DNA binding protein (TARDBP), FUS RNA binding protein (FUS), Optineurin (OPTN), valosin containing protein (VCP), Ubiquilin 2 (UBQLN2), chromosome 9 open reading frame 72 (C9ORF72), and profilin 1 (PFN1) (8). In 2014, Chen and colleagues generated iPSCs from individuals with SOD1 mutations to investigate early pathological events in motor neurons derived from these iPSCs. In their study, they showed that SOD1-mutant motor neurons exhibited neurofilament aggregation and altered stoichiometry for the neurofilament subunits (9). In the same year, Eggan et al. demonstrated transcriptional changes in motor neurons derived from the iPSCs of individuals with ALS caused by pathogenic repeat expansions in the C9orf72 locus, suggesting that these distinct disease-causing mutations act through common pathways (10).
Summary
Human neural progenitor cells (hNPCs) have the potential to be therapeutically beneficial in the treatment of neuronal diseases. The direct generation of specific subtypes of neurons, astrocytes, and oligodendrocytes from human fibroblast or iPSCs and ESCs can provide us with a lot more information on genetic variation, disease-causing mutations, and their relevance in affecting neural development.
Written by Karolina Szczesna; PhD in Biomedicine
Product Manager, Proteintech Ltd.
References
1. Alzheimer mechanisms and therapeutic strategies.
3. Modeling familial Alzheimer's disease with induced pluripotent stem cells.
5. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells.
6. Parkinson's disease gene therapy: success by design meets failure by efficacy.
7. Parkinson's disease induced pluripotent stem cells with triplication of the α-synuclein locus.
8. State of play in amyotrophic lateral sclerosis genetics.
10. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons.
Related Content
Proteintech interview with an early-career researcher: Odetta Antico
The Neurochemistry of Parkinson's Disease

Support
Newsletter Signup
Stay up-to-date with our latest news and events. New to Proteintech? Get 10% off your first order when you sign up.