
Human/Mouse SMN ELISA Kit
Cat no : KE00027
Synonyms
Component of gems 1, Gemin 1, SMN, SMN1, SMN2, SMNC, SMNT, Survival motor neuron protein
Validation Data Gallery
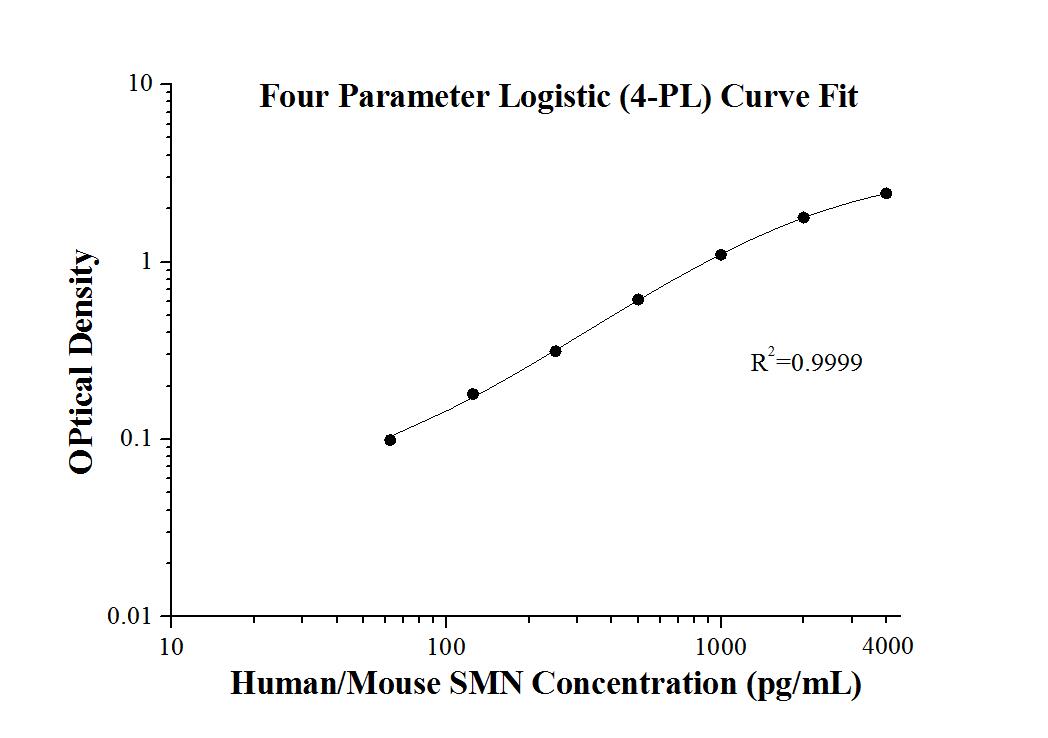
Product Information
KE00027 is a solid phase sandwich Enzyme Linked-Immuno-Sorbent Assay (Sandwich ELISA). The SMN ELISA kit is designed for the quantitative determination of SMN in samples of human and mouse origin. An antibody specific for SMN has been pre-coated onto the microwells. The SMN protein in samples is captured by the coated antibody after incubation. Following extensive washing, another antibody specific for SMN is added to detect the captured SMN protein. For signal development, horseradish peroxidase (HRP)-conjugated antibody is added, followed by Tetramethyl-benzidine (TMB) reagent. Solution containing sulfuric acid is used to stop color development and the color intensity which is proportional to the quantity of bound protein is measurable at 450 nm with the correction wavelength set at 630 nm.
| Product name | Human/Mouse SMN ELISA Kit |
| Tests | 1 X 96 well plate |
| Sample type | Cell lysates, Tissue lysates |
| Assay type | Sandwich |
| Sensitivity | 6.0 pg/mL |
| Range | 62.5-4000 pg/mL |
| Reactivity | Human/Mouse |
| Tested applications | Sandwich ELISA |
| Gene ID (NCBI) | 6607 |
Recovery
| Sample Type | Average | Range |
|---|---|---|
| Cell lysate | 101% | 83%-127% |
| Tissue lysate | 93% | 71%-108% |
IntraAssay
| Sample | n | mean ( pg/mL) | SD | CV% |
|---|---|---|---|---|
| 1 | 20 | 1,037.6 | 63.2 | 6.1 |
| 2 | 20 | 1,678.6 | 71.1 | 4.2 |
| 3 | 20 | 2,465.5 | 98.0 | 4.0 |
InterAssay
| Sample | n | mean ( pg/mL) | SD | CV% |
|---|---|---|---|---|
| 1 | 24 | 1,219.4 | 93.9 | 7.7 |
| 2 | 24 | 2,594.4 | 152.6 | 5.9 |
| 3 | 24 | 4,296 | 252.7 | 5.9 |
Background Information
Survival Motor Neuron (SMN) protein required for efficient assembly of small nuclear ribonucleoprotein (snRNP) complexes is encoded by nearly identical telomeric and centromeric forms of SMN gene respectively. Both the SMN1 and SMN2 genes express SMN protein, however, the amount of functional full-length protein produced by SMN2 is much less than that produced by SMN1 due to the alternative splicing.The SMN gene is constitutively expressed in wide variety of tissues including brain, kidney, liver and spinal cord, while motor neurons are particularly vulnerable to reduced SMN protein levels. Deletion or mutational inactivation of the SMN1 gene causes spinal muscular atrophy (SMA), a lethal genetic disease characterized by loss of motor neurons in the spinal cord. The absence of SMN1 can be partially compensated for by SMN2 and the SMN2 expression level is associated with SMA severity. This kit is for the quantitative determination of SMN protein level in vivo.
Properties
| Storage Instructions | All the reagents are stored at 2-8℃ for 6 months or -20℃ for 12 months. Refer to the protocol for further storage instructions. |
| Synonyms | Component of gems 1, Gemin 1, SMN, SMN1, SMN2, SMNC, SMNT, Survival motor neuron protein |
FAQ
1、I have limited sample diluent. How should I handle samples requiring high dilution factors?
Answer: We recommend performing a serial dilution. For example, if human serum requires a 1,000,000-fold dilution, you can use a three-step 100× dilution method as follows:
Step 1: Take 2 uL of human serum and add it to 198 uL of sample diluent, mix thoroughly to obtain a 100-fold diluted sample.
Step 2: Take 2 uL of the 100-fold diluted sample and add it to 198 uL of sample diluent, mix thoroughly to obtain a 10,000-fold diluted sample.
Step 3: Take 2 uL of the 10,000-fold diluted sample and add it to 198 uL of sample diluent, mix thoroughly to obtain a 1,000,000-fold diluted sample.
If further dilution is required, additional serial steps can be used. We do not recommend using any other solutions or water for dilution, except for the sample diluent we suggest. If your sample diluent is still insufficient, please contact our customer service for more sample diluent.