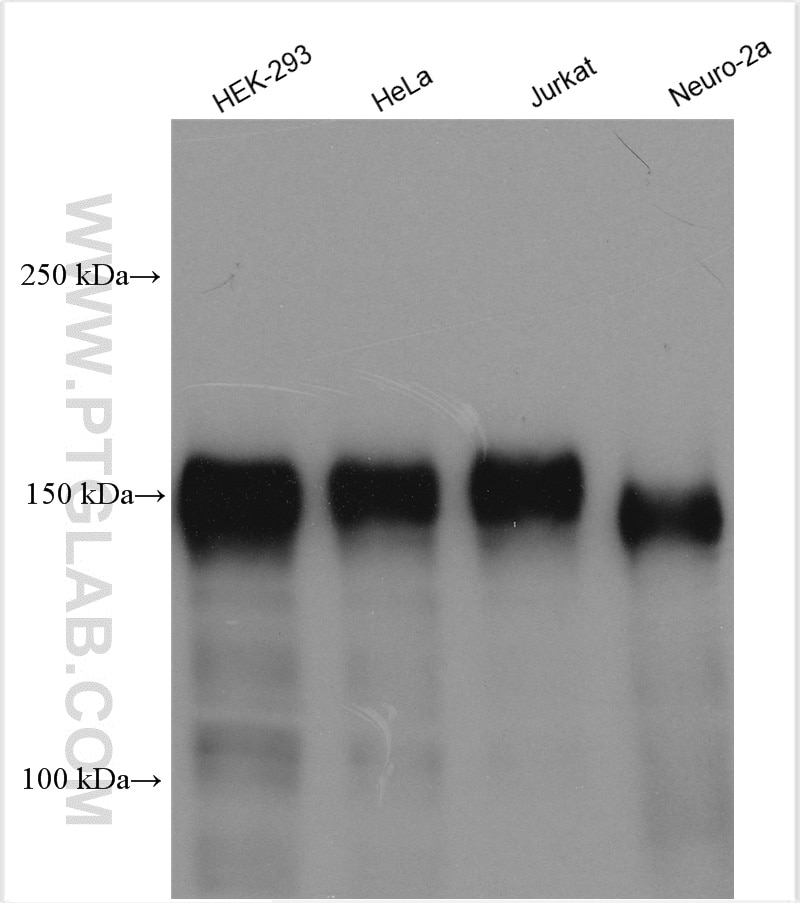
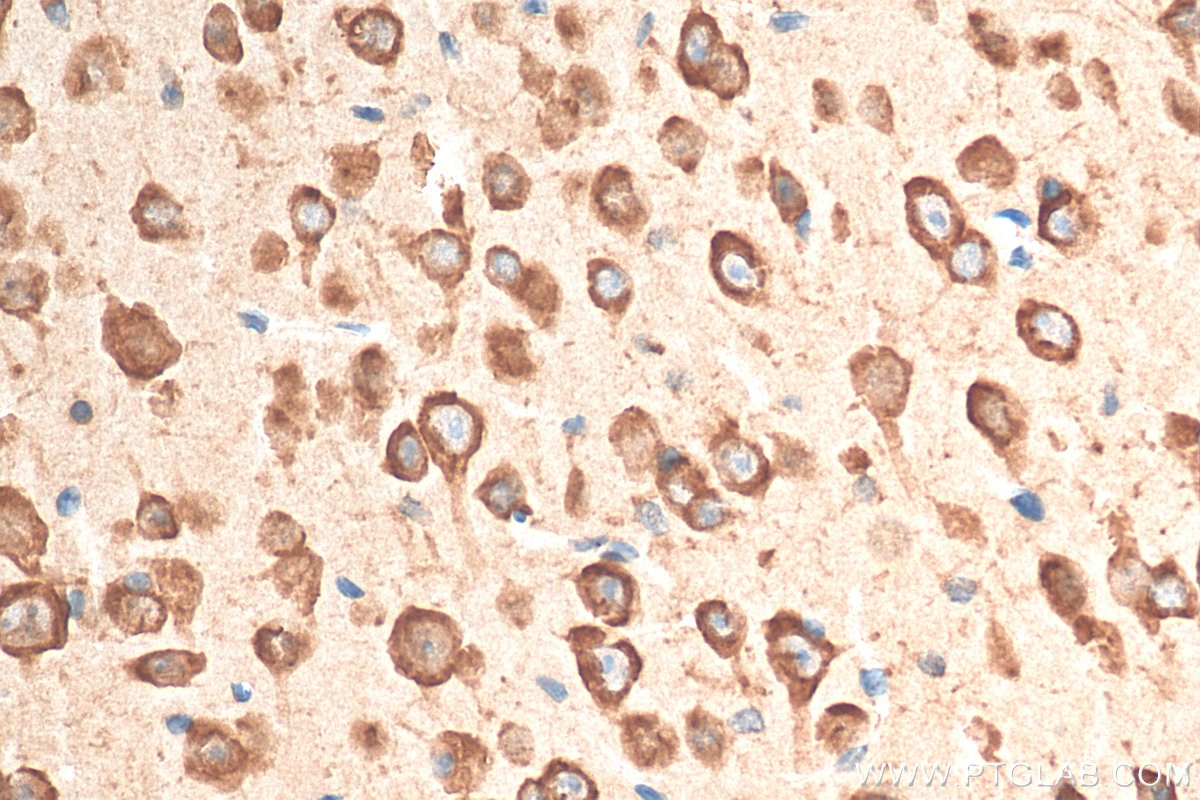
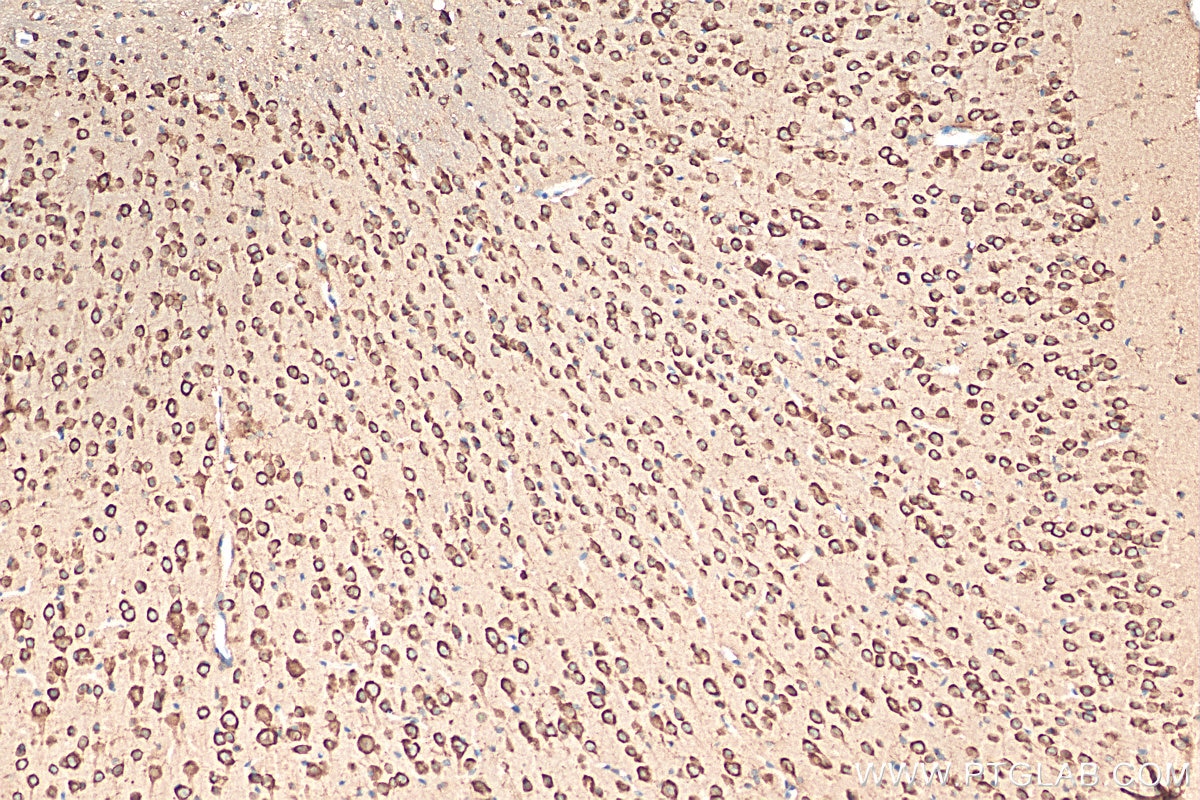
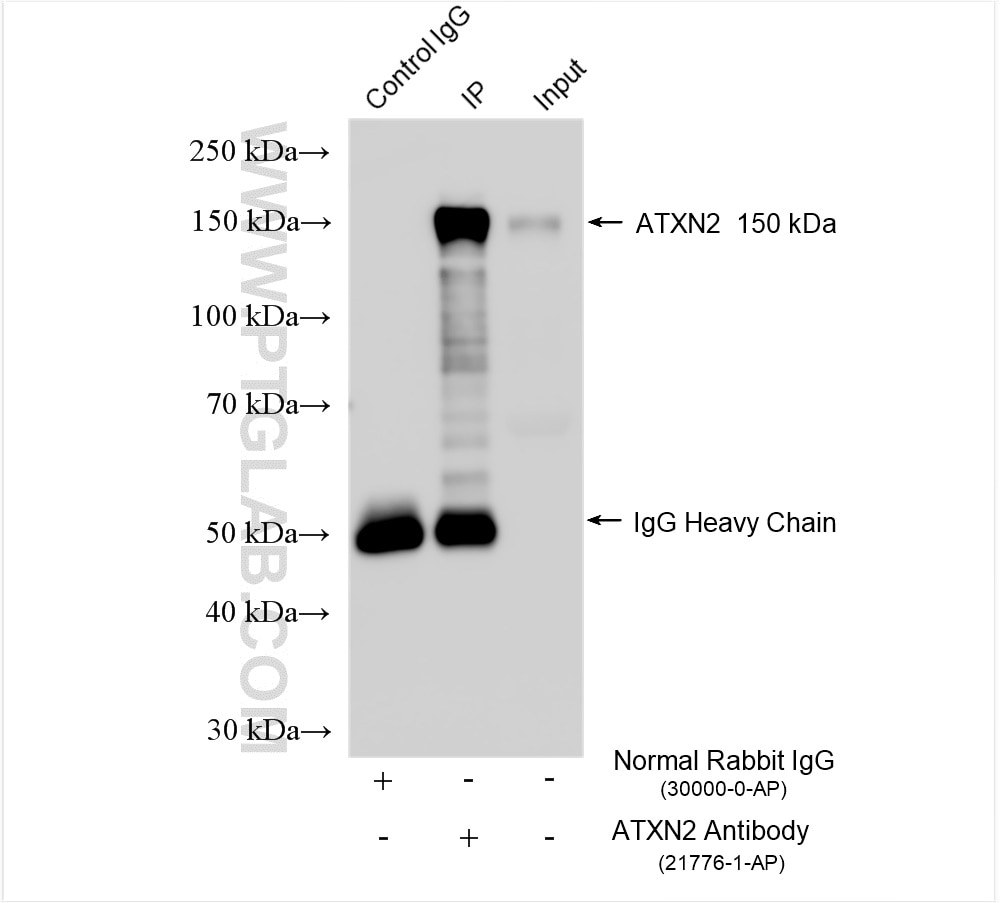
Tested Applications
| Positive WB detected in | HeLa cells, HEK-293 cells, Jurkat cells, Neuro-2a cells |
| Positive IP detected in | HEK-293 cells |
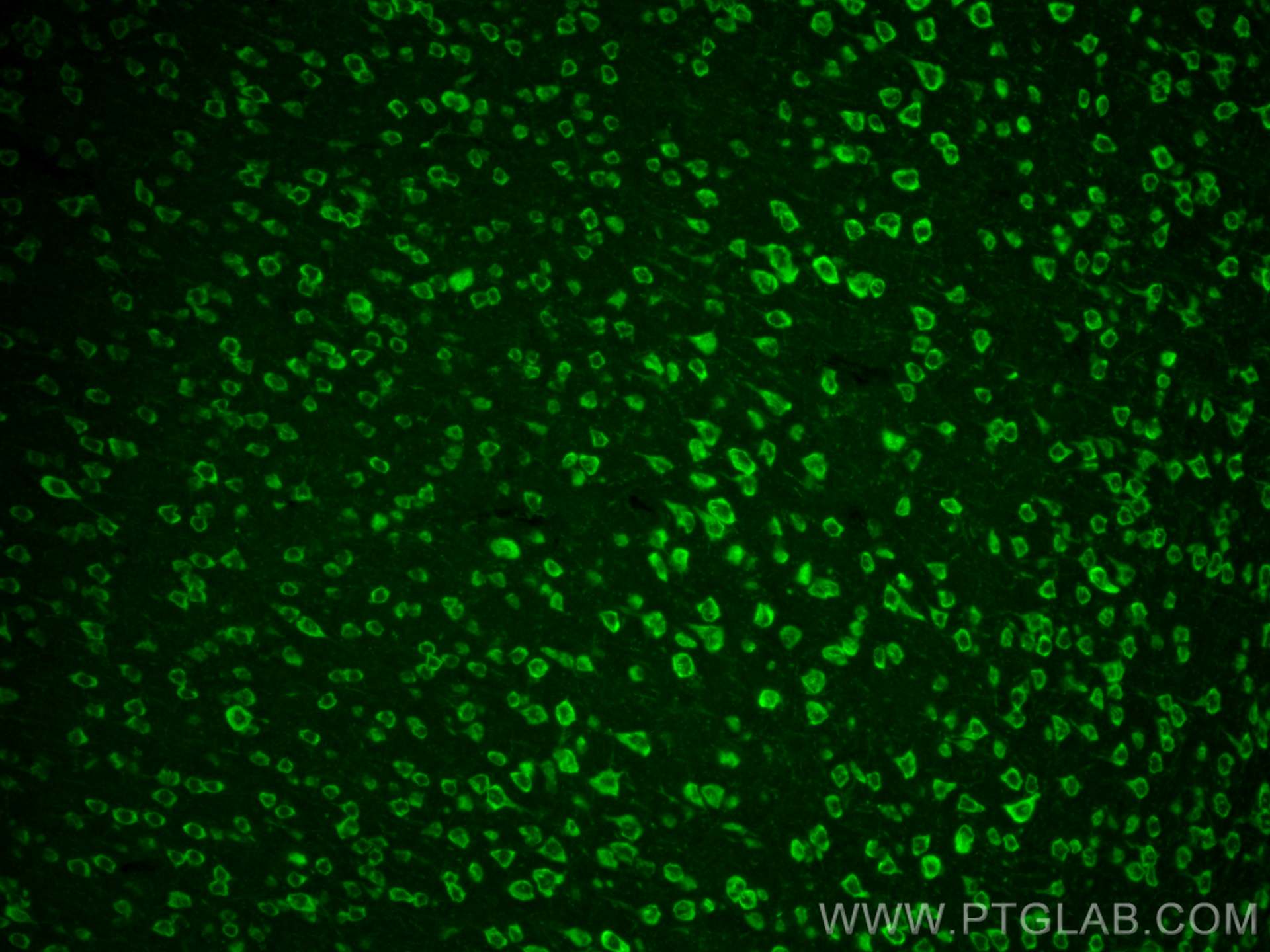
| Positive IHC detected in | mouse brain tissue Note: suggested antigen retrieval with TE buffer pH 9.0; (*) Alternatively, antigen retrieval may be performed with citrate buffer pH 6.0 |
| Positive IF-P detected in | mouse brain tissue |
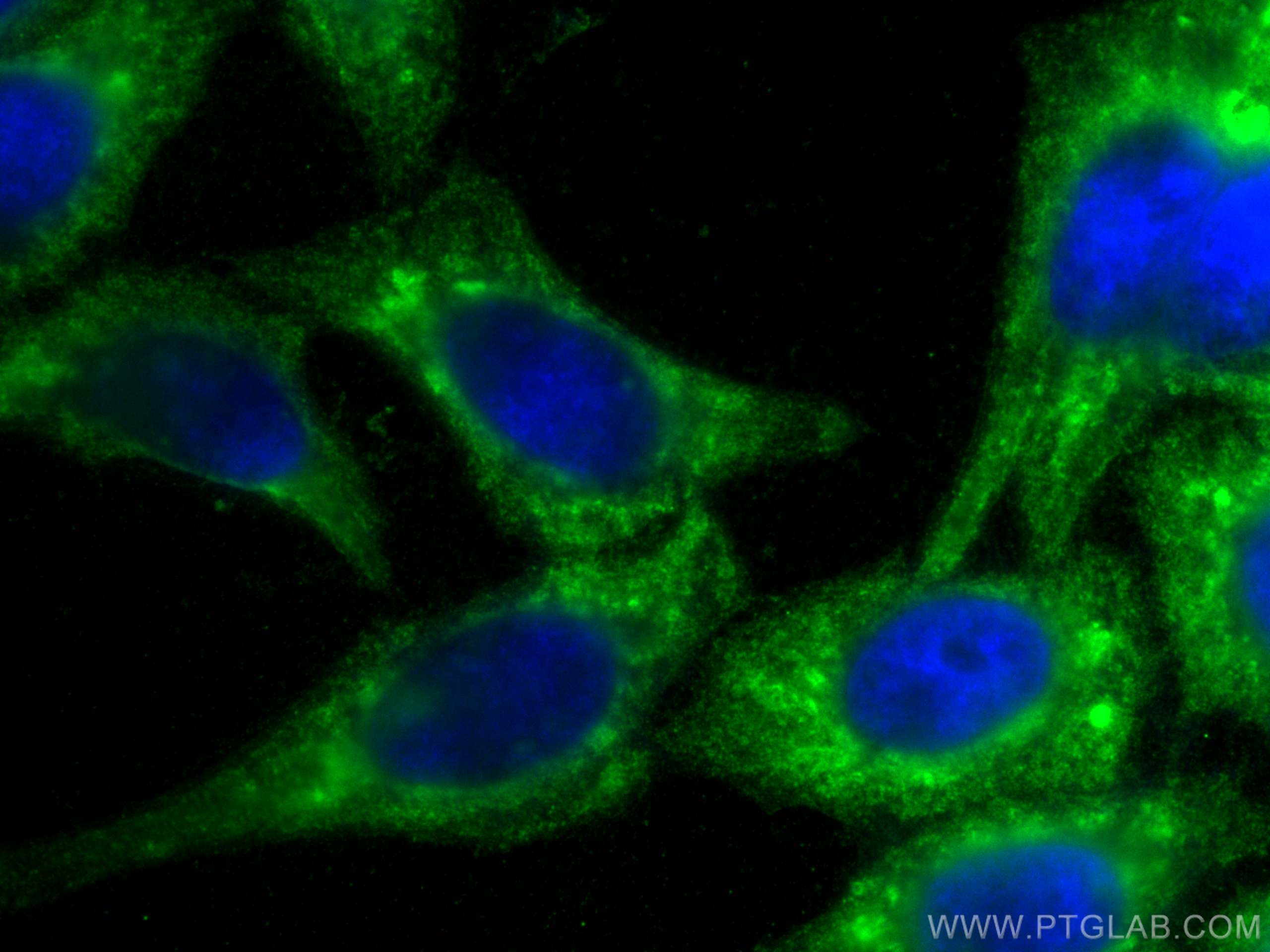
| Positive IF/ICC detected in | HepG2 cells |
Recommended dilution
| Application | Dilution |
|---|---|
| Western Blot (WB) | WB : 1:2000-1:16000 |
| Immunoprecipitation (IP) | IP : 0.5-4.0 ug for 1.0-3.0 mg of total protein lysate |
| Immunohistochemistry (IHC) | IHC : 1:50-1:500 |
| Immunofluorescence (IF)-P | IF-P : 1:50-1:500 |
| Immunofluorescence (IF)/ICC | IF/ICC : 1:450-1:1800 |
| It is recommended that this reagent should be titrated in each testing system to obtain optimal results. | |
| Sample-dependent, Check data in validation data gallery. | |
Published Applications
| KD/KO | See 5 publications below |
| WB | See 26 publications below |
| IHC | See 3 publications below |
| IF | See 19 publications below |
| IP | See 4 publications below |
| CoIP | See 1 publications below |
Product Information
21776-1-AP targets Ataxin 2 in WB, IHC, IF/ICC, IF-P, IP, CoIP, ELISA applications and shows reactivity with human, mouse, rat samples.
| Tested Reactivity | human, mouse, rat |
| Cited Reactivity | human, mouse, rat |
| Host / Isotype | Rabbit / IgG |
| Class | Polyclonal |
| Type | Antibody |
| Immunogen |
CatNo: Ag16470 Product name: Recombinant human Ataxin 2 protein Source: e coli.-derived, PGEX-4T Tag: GST Domain: 251-600 aa of BC114546 Sequence: GSDQRVVNGGVPWPSPCPSPSSRPPSRYQSGPNSLPPRAATPTRPPSRPPSRPSRPPSHPSAHGSPAPVSTMPKRMSSEGPPRMSPKAQRHPRNHRVSAGRGSISSGLEFVSHNPPSEAATPPVARTSPSGGTWSSVVSGVPRLSPKTHRPRSPRQNSIGNTPSGPVLASPQAGIIPTEAVAMPIPAASPTPASPASNRAVTPSSEAKDSRLQDQRQNSPAGNKENIKPNETSPSFSKAENKGISPVVSEHRKQIDDLKKFKNDFRLQPSSTSESMDQLLNKNREGEKSRDLIKDKIEPSAKDSFIENSSSNCTSGSSKPNSPSISPSILSNTEHKRGPEVTSQGVQTSS Predict reactive species |
| Full Name | ataxin 2 |
| Calculated Molecular Weight | 1313 aa, 140 kDa |
| Observed Molecular Weight | 140-150 kDa |
| GenBank Accession Number | BC114546 |
| Gene Symbol | ATXN2 |
| Gene ID (NCBI) | 6311 |
| RRID | AB_10858483 |
| Conjugate | Unconjugated |
| Form | Liquid |
| Purification Method | Antigen affinity purification |
| UNIPROT ID | Q99700 |
| Storage Buffer | PBS with 0.02% sodium azide and 50% glycerol, pH 7.3. |
| Storage Conditions | Store at -20°C. Stable for one year after shipment. Aliquoting is unnecessary for -20oC storage. 20ul sizes contain 0.1% BSA. |
Background Information
Background
ATXN2 (Ataxin-2) is a eukaryotic RNA‐binding protein that is conserved across species. It contains two LSm domains (RNA binding), a PAM2 motif (association with poly(A)-binding protein), and an N-terminal polyglutamine tract (PMID: 25027299). Ataxin-2 plays crucial roles at different stages of the regulation of posttranslational gene expression. ATXN2 is also directly implicated in the regulation of neural function by influencing specific molecular and cellular pathways. Genetic expansion of a poly‐glutamine tract in human ATXN2 has been linked to several neurodegenerative diseases, where it most likely acts through gain‐of‐function effects.
What is the molecular weight of ATXN2?
The molecular weight varies from 12 to 140 kDa, depending on the isoform.
What are the isoforms of ATXN2?
ATXN2 has many isoforms, which differ significantly in length and domain composition.
What is the subcellular localization of ATXN2?
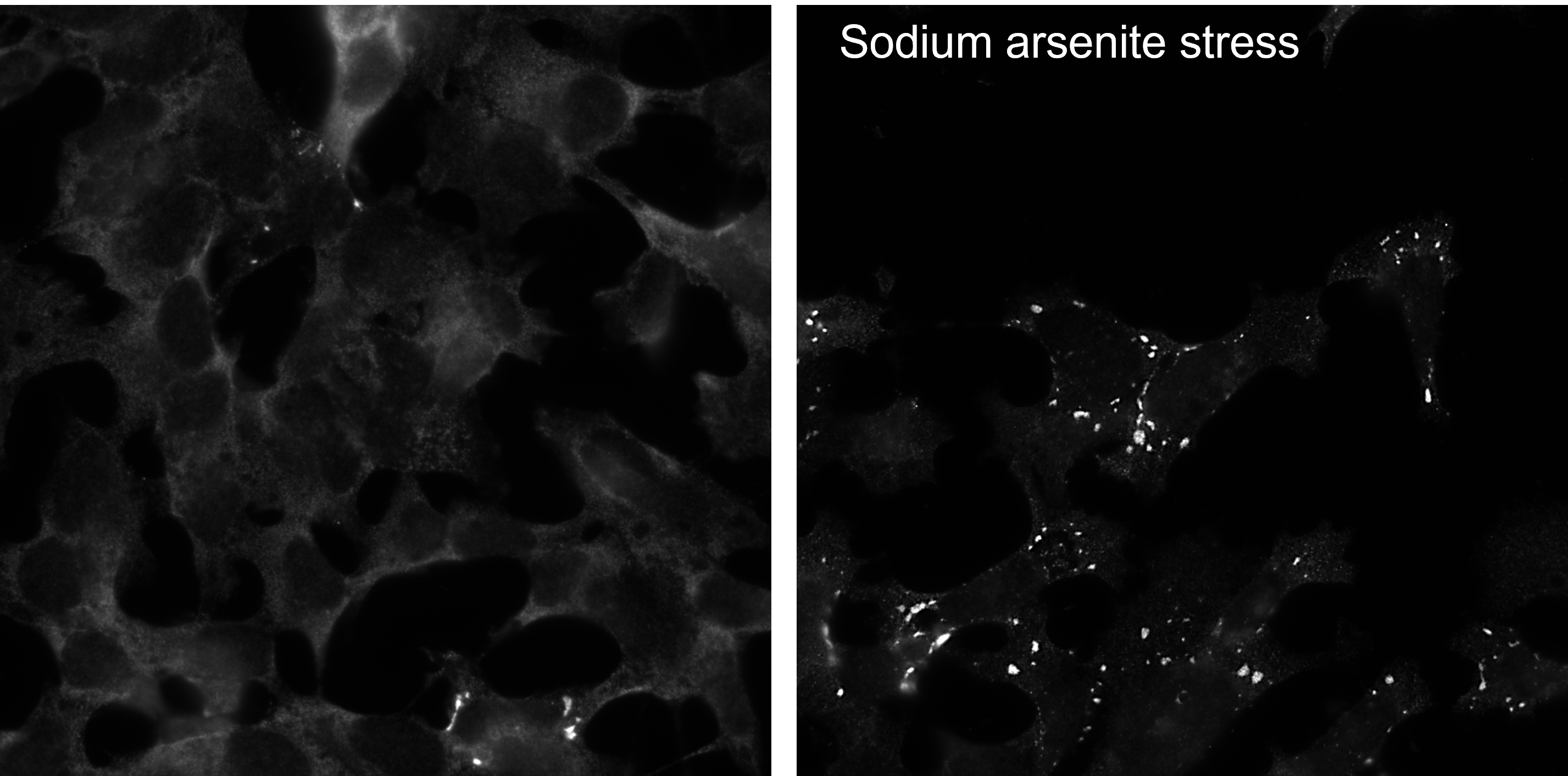
According to siRNA-based experiments, it is localized in the cytoplasmic compartment of normal cells, where it mostly associates with the Golgi apparatus and stress granules (PMID: 22508507).
What is the tissue specificity of ATXN2?
ATXN2 protein has been detected in various tissues; however, it is most highly expressed and studied in CNS. Significant amounts of this protein can also be detected in the liver and gallbladder.
What is the molecular function of ATXN2?
Ataxin-2 is involved in regulating various steps of mRNA translation, including poly‐A tailing, RNA stabilization, microRNA‐dependent gene silencing, and translational activation. All those functions are linked to its interactions with the poly(A)-binding protein. Ataxin-2 is involved in the formation of stress granules and P-bodies. Furthermore, genetic models of ATXN2 loss‐of‐function have underlined the importance of ATXN2 in mTOR signaling and cellular metabolism, which are all crucial for neural homeostasis (PMID: 29869836).
What is ATXN2's involvement in disease?
The N-terminal region of ATXN2 normally contains a polyQ stretch of 14-31 residues that can be expanded in the pathogenic state to 32-200 residues. Intermediate length expansions of this tract increase susceptibility to amyotrophic lateral sclerosis (ALS or Lou Gehrig's disease) (PMID: 20740007). Long expansions of this tract result in spinocerebellar ataxia-2 (SCA2), an autosomal dominantly inherited, neurodegenerative disorder (PMID: 29427103). Genome-wide association studies indicate that loss-of-function mutations in this gene are associated with susceptibility to type I diabetes, obesity, and hypertension.
Protocols
| Product Specific Protocols | |
|---|---|
| IF protocol for Ataxin 2 antibody 21776-1-AP | Download protocol |
| IHC protocol for Ataxin 2 antibody 21776-1-AP | Download protocol |
| IP protocol for Ataxin 2 antibody 21776-1-AP | Download protocol |
| WB protocol for Ataxin 2 antibody 21776-1-AP | Download protocol |
| Standard Protocols | |
|---|---|
| Click here to view our Standard Protocols |
Publications
| Species | Application | Title |
|---|---|---|
Cell Diverse CMT2 neuropathies are linked to aberrant G3BP interactions in stress granules | ||
Mol Cell Autism-Misregulated eIF4G Microexons Control Synaptic Translation and Higher Order Cognitive Functions. | ||
Neuron Small-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD. | ||
Sci Adv Endogenous retrovirus-like proteins recruit UBQLN2 to stress granules and shape their functional biology | ||
Nat Commun NUP62 localizes to ALS/FTLD pathological assemblies and contributes to TDP-43 insolubility. |
Reviews
The reviews below have been submitted by verified Proteintech customers who received an incentive for providing their feedback.
FH Tatyana (Verified Customer) (01-21-2023) | Very good clean signal in ICC. Labels cytoplasmic stress granules well. Antibody was incubated 2 h at RT at 1:100o dilution and could also be reused.
 |