
Mass spec essentials
An overview of mass spec sample preparation, sample purification and data analysis.
Mass spectrometry (mass spec; MS) is a powerful method for analyzing macromolecules.
Mass spec dependent on their mass (m) and charge (z) after ionization from pure or more complex sample mixtures.
Thanks to rapid advances in both technical instruments and analytical software, the range and versatility of applications have increased significantly during the 21st century. Although mass spec is still frequently used for “simple” compound or peptide identification, recent approaches are more focused on the targeted analysis of bigger datasets.
Mass spectrometry can be divided into 3 steps:
1. Sample preparation
2. Sample purification
3. Data analysis (Figure 1)
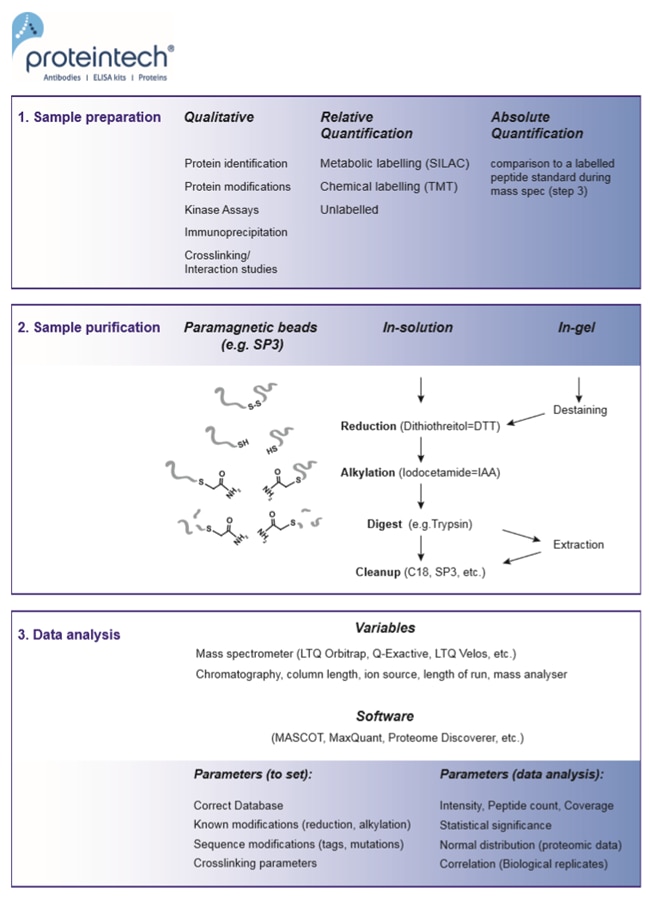
Fig. 1. Mass spec in 3 steps: sample preparation, sample purification, and data analysis.
1. Sample preparation
Abundance of proteins
Mass spectrometry is among the most sensitive protein detection methods, capable of detecting proteins in the femto-range.
However, there are situations in which these capabilities may be compromised and require monitoring and validation. If intracellular protein concentration is low, the protein is prone to degradation or occupies an intracellular compartment, more abundant cytosolic proteins can mask the signal from the protein of interest in complex sample sets during detection.
To anticipate any problems before subjecting the sample to mass spectrometry, the experiment should be monitored throughout by SDS PAGE, or preferably via Western blot.
To enrich for low-abundant proteins, one may consider immunoprecipitation or cell fractionation. If cell fractionation is performed, Western blots should be probed in parallel for cell organelle markers to ensure proper separation of cell compartments.
Controls
The necessary set of controls is dependent on the experiment.
Qualitative experiments:
Examples are immunoprecipitations or kinase assays. The usual biological controls will work here, e.g., “no antibody,” “no tag,” “no kinase,” “wildtype versus mutant.”
Relative quantification:
This includes metabolic labelling (SILAC: Stable Isotope Labelling by Amino acids in Cell culture) as well as chemical labelling (TMT: Tandem Mass Tags). See Figure 2 below for an overview of these methods. Differences in peptide abundance between up to 3 (SILAC) or 10 samples (TMT) can be monitored. The use of several isotope-labelled amino acids is crucial for verification of the resulting datasets. This means analyzing a biological replicate with inverse labelling of the samples in order to exclude systematic errors. The least expensive but also least accurate version is label-free quantification, where spectra of two separate runs are compared via AUC (area under the curve) or spectral counting.
Absolute quantification (AQUA):
A labelled synthetic peptide of known concentration is “spiked in” during MASS spec analysis of the sample. Consequently, this is a highly precise method. However, due to synthesis of the labelled peptide, it can be quite expensive.
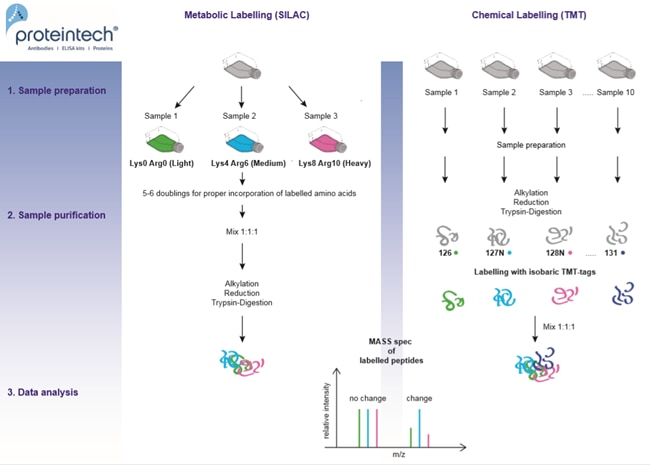
Fig. 2. Relative quantification methods: metabolic versus chemical labelling.
Stability of protein and its modifications
If there is the possibility of proteases (or phosphatases) in the mixture, remember to include sufficient inhibitors in the sample mixture.
During the experiment, it is recommended to perform as many steps as possible at low temperature (4°C) to minimize proteolytic activity. Protease inhibitors should be depleted from the mixture before sample preparation for mass spec analysis, as they interfere with trypsin digestion (or any other protease used).
Contaminants
All surfaces, containers, tips, and solutions should be clean to avoid keratin contamination, which is highly abundant and can mask the signal.
Polymers, especially PEG (Polyethylene glycol), can also be a source of contamination. PEG can derive from detergents used for cleaning glassware or can be released from plastic containers and tips during autoclaving.
It is best to use HPLC-grade water for all working solutions, non-autoclaved filter tips, single-use pipettes, and low-protein binding tubes. In some cases, polymer contamination can also derive from affinity column material. In that case, consider running your sample on a gel before trypsin digestion and extraction as an additional cleaning step. Pre-cast gels are preferable, as they present less risk of keratin contamination. For self-cast gels, use HPLC-grade water and clean all material thoroughly with ethanol.
2. Sample purification
Enzyme digestion
After reduction and alkylation, proteins must be digested into peptides of the right size. The optimal peptide length for detection is 8 – 15 amino acids (1).
The most commonly used enzyme for digestion is trypsin. Please note: peptide lengths after digestion can be predicted online with freely available PeptideMass software (https://web.expasy.org/peptide_mass/). If there are not enough or too many trypsin recognition sites, other proteases can be used to generate a more suitable range of peptides.
In-gel or in-solution digestion
Proteins can be digested either directly in solution or after they are run on a gel, followed by extraction of the digested peptides.
| In solution | |
| Advantage | No sample material loss |
| Disadvantage | Buffer must be compatible with the trypsin digestion |
| In gel | |
| Advantage | Additional cleaning step to avoid contaminants like polymers; Further separation of proteins according to their sizes by cutting the lane into different fractions. |
| Disadvantage | Loss of sample material. |
Cleaning up
To remove contaminants, salts, and buffers, clean-up is a necessary last step before subjecting the sample to mass spectrometry. Most commonly used are C18 columns (reversed phase resin), which bind the hydrophobic peptides, so that salts and hydrophilic contaminants can be washed out.
Next, peptides can be eluted with organic solvents. More recent methods like SP3 (Single-Pot Solid-Phase-enhanced Sample Preparation) bind proteins to magnetic beads where clean-up and digestion can be performed directly, enabling more stringent washes, minimizing the loss of material and also increasing the reproducibility (2).
3. Data analysis
Database
- Choose the protein database that is appropriate for your experiment and organism from UniProt (https://www.uniprot.org) and load the latest version into your MASS spec software.
- If one of the proteins contains an epitope-tag, be sure to include the tagged sequence of the protein in the database you are using for data analysis.
Reversible crosslinkers
- Before sample purification, the crosslinking reagent is chemically cleaved, leaving the interacting proteins with a traceable modification. Include this modification in your database.
Non-reversible crosslinkers
- Use specialized software and guidance to interpret the data, as crosslinked peptides can derive from different proteins. That adds an additional layer of complexity to the data analysis.
Reproducibility
- If analyzing whole proteome datasets, ensure that the entity of all peptides detected in one experiment follows a Gaussian distribution.
- Datasets obtained from different biological replicates must agree with each other. Otherwise, the results are invalid.
- The result/identification of the peptide must be statistically significant. The p-value (also Q-value or Score, dependent on software) should be < 0.05.
References
1. The value of using multiple proteases for large-scale mass spectrometry-based proteomics.
2. Ultrasensitive proteome analysis using paramagnetic bead technology.
Able™