
Quantitative MiRNA Profiling: How To Choose The Best RNA Sequencing Method?
RNA sequencing (RNA-seq) is a widely used method employing next generation sequencing (NGS) to discover and quantify the abundance of RNA molecules in biological samples.
Why do we do RNA-seq?
RNA-seq has revolutionized approach to transcriptomics, it allows high-throughput being precise and cost-effective analysis of RNAs having advantages over microarrays and expressed sequence tag (EST) sequencing [1] (Table 1).
|
Parameter |
DNA microarrays |
EST sequencing |
RNA-seq |
|
Detection method |
Hybridization |
Sanger sequencing |
NGS |
|
Sensitivity |
Yes, but with the limitation –detect changes in expression levels up to 10,000-fold |
Limited – cannot analyze small RNAs (<500 bp); not always is possible to map reads for RNA specific isoforms |
Yes – possible to detect low expressed RNAs by increased sequencing depth; single base resolution |
|
Amount of RNA needed |
High/medium – 1-10 ng |
High – requires cDNA libraries |
Low -even single cell analysis possible |
|
Costs |
Medium |
High/medium – mostly associated with cDNA libraries and Sanger sequencing cost |
Low |
RNA-seq technology has many applications in basic and clinical research (Figure 1). It is mainly used for:
- functional annotation of genomes by identifying all coding and non-coding RNAs, splice-sites, isoforms, open reading frames
- quantitative analysis of RNA expression and expression profiling
- detection of single nucleotide polymorphisms
- studying RNA editing – RNA posttranscriptional modifications (PTMs)
Specialized RNA-seq setups can focus on subsets of RNAs, e.g. coding RNAs (mRNA sequencing and exome capture) or ribosomal RNAs (ribosome profiling). Increased sensitivity of RNA detection led to development of sequencing single cells (single cell RNA-seq), which is invaluable tool for studying tissues complexity or heterogeneous cell populations on a single-cell level.
RNA-seq was developed to determine RNA structure (selective 2′-hydroxyl acylation analyzed by primer extension - SHAPE) and map RNA-RNA and RNA-protein interaction sites (cross-linking immunoprecipitation - CLIP).
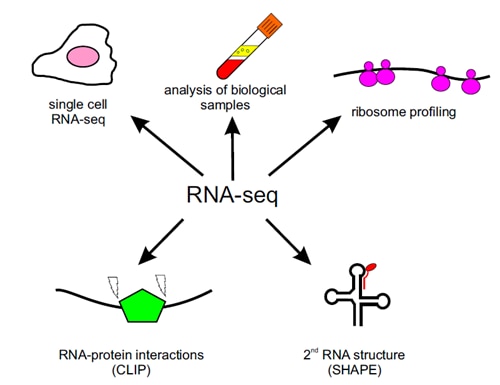
Figure 1. RNA-seq technology applications in basic and clinical research.
Challenges in small RNA-seq
Developed just 10 years ago, RNA-seq was firstly used for poly-A containing messenger RNAs [2-4]. Standard RNA-seq (“long RNA-seq”) is used for sequencing messenger RNAs and long non-coding RNAs. It is based on RNA isolation followed by its fragmentation to the desired length 30–400 bp next reversely transcribed to cDNA. On the contrary, small RNAs, such as mature microRNAs (miRNAs) that are ~22 nucleotides are too small for standard RNA-seq protocols. Due to the small length of miRNAs, additional steps of extension (such as RNA ligation or poly-A tailing) during library preparation are needed to allow unbiased and efficient reverse transcription and Polymerase Chain Reaction (PCR) amplification (“small RNA-seq”, Figure 2). A proper normalization and reproducibility assessment are Needed in particular for biological samples with low RNA abundance such as examination of circulating cell-free miRNAs in the blood. Giraldez et al. Nature Biotechnology [5] benchmarked nine different library preparation methods assessing their reproducibility, accuracy and ability to detect miRNA editing.
This was done by using three reference samples:
1) an equimolar pool of over 1,000 short synthetic RNAs
2) pools of small synthetic RNAs mixed in different molar ratios
3) a small RNA pool isolated from human blood.
Nine independent research groups prepared and analyzed libraries in replicates, which resulted in an impressive number of 364 RNA-seq datasets.
Different methods of small RNA-seq
Several commercial and in-house methods have been developed for performing small RNA-seq experiments. Most available methods require 3’ and 5’ adapter ligations. This is a crucial step that has a direct impact on the quality and selectivity of amplified RNAs. It has been demonstrated that RNA ligases (Rnl1 and Rnl2) used in ligation steps have sequence and structural biases that lead to overrepresentation and underrepresentation of certain RNA subsets [6, 7]. Current protocols and strategies for small RNA-seq can be divided into two categories: those employing adapters with invariant ends, where adapters have strictly predefined sequences, and those with adapters containing four degenerate nucleotides at the ligation ends (4N adapters) (Figure 2).
Giraldez et al. evaluated three commercial kits using invariant ends: TruSeq (Illumina), NEBNext (New England BioLabs) and CleanTag (Trilink Biotech), and six protocols with 4N adapters: commercial NEXTflex (Bioo Scientific), a recently published protocol by Ping and collegues [8] and four in-house protocols.
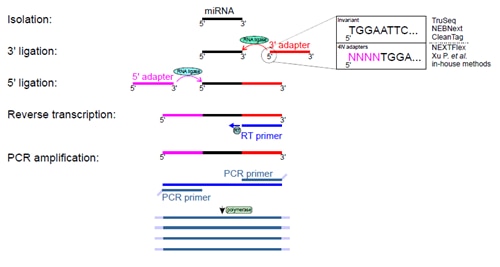
Figure 2. Experimental steps of small RNA-seq. Isolated small RNAs undergo two rounds of ligations with 3’ and 5’ adapters and these reactions are catalyzed by two RNA ligases. Obtained products serve as templates for reverse transcription and DNA fragments are amplified by a few cycles of PCR. Amplified DNA products undergoes sequencing. Giraldez et al. [5] compared nine protocols that use 3’ and 5’ adapters either with invariant ends or adapters with 4 four degenerate nucleotides at their ligation ends. The figure depicts variants of 3’ adapter ends only for simplicity.
1st sequence-bias is a real problem
Giraldez et al. [5] reports that currently used small RNA-seq protocols suffer from sequence bias. Observed sequence bias between different protocols was significantly higher than between datasets generated by different research groups using the same protocol, suggesting that the choice of protocol for RNA-seq has a major impact on the quality of obtained data. This should be kept in mind while comparing small RNA-seq results of similar biological phenomena that were analyzed using different library preparation protocols. 4N adapter raised considerably smaller sequence bias (2-20 times protocol depending) than invariant adapter methods and therefore is a preferred choice. Reduced sequence bias in 4N adapter protocols also suggest they should perform better while detecting variety of small RNA species (sample coverage), even if the sequencing has lower depth.
2nd small RNA-seq protocols are accurate and reproducible
The use of predefined pools of small synthetic RNAs allowed comparison of all protocols in terms of their accuracy and reproducibility between different research groups. Observed miRNA abundance ratios were very close to expected ratios, which mean all protocols were accurate for obtaining correct relative miRNA abundance. Reproducibility was measured in synthetic and biological small RNA samples by statistical parameters (coefficient of variation, quartile coefficient of dispersion). Detailed results are summarized in supplementary data set (the coefficient variation for most of identified RNAs was below 20%) [5].
3rd detecting miRNA is most reliable with an in-house 4N adapter protocol
Deamination of adenosine residues to inosine (A-to-I editing) can occur in a subset of miRNA precursors. It has an impact on their further processing and biological activity [9]. Therefore, it is important that researchers use appropriate RNA-seq method that allows precise detection of A-to-I miRNA editing. While using a reference pool of edited and unedited small synthetic RNAs mixed in different molar ratios, Giraldez et al. [5] has evaluated that three out of nine protocols allow detecting A-to-I miRNA editing. All examined protocols had a good specificity (>99%) to reliably detect and discriminate between edited and unedited RNAs in samples. However, an in house 4N adapter protocol performed better than commercial invariant adapter protocols in accurately detecting actual editing levels. This is most likely due to the fact that 4N adapter protocols having lower sequence bias maximizing sequence coverage and therefore outperforming invariant adapter protocols.
Further improvements needed…
Without any doubt, this comprehensive study helps researchers follow-up more deeply regarding protocols and experimental designs details for small RNA-seq. Small RNA-seq still displays large sequence biases when compared with long RNA-seq. Importantly, recent investigations of small RNA-seq analysis that have used 4N adapters seem to yield a lower sequence bias. It is worth mentioning that the four studied 4N adapter protocols generated datasets of different quality, suggesting that additional parameters such as ligation times and temperatures should be taken into account while performing experiments regarding small RNA-seq.
Based on:
Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling Nature Biotechnology volume 36, pages 746–757 (2018)
Posted by Dr. Karolina Szczesna, Senior Product Manager and Technical Support at Proteintech Ltd.
References list
- Wang, Z., M. Gerstein, and M. Snyder, RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet, 2009. 10(1): p. 57-63.
- Mortazavi, A., et al., Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods, 2008. 5(7): p. 621-8.
- Nagalakshmi, U., et al., The transcriptional landscape of the yeast genome defined by RNA sequencing. Science, 2008. 320(5881): p. 1344-9.
- Wilhelm, B.T., et al., Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature, 2008. 453(7199): p. 1239-43.
- Giraldez, M.D., et al., Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat Biotechnol, 2018. 36(8): p. 746-757.
- Jayaprakash, A.D., et al., Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Res, 2011. 39(21): p. e141.
- Zhuang, F., et al., Structural bias in T4 RNA ligase-mediated 3'-adapter ligation. Nucleic Acids Res, 2012. 40(7): p. e54.
- Ping, X., et al., An improved protocol for small library construction using High Definition adapters. Methods Next-Generation Seq., 2015(2): p. 1-10.
- Yang, W., et al., Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol, 2006. 13(1): p. 13-21.
Able™