
8 Top Tips For Immunoprecipitation
8 tips on how to perform better IP experiments.
Introduction
Immunoprecipitation, or IP, is a well-established technique for the isolation of proteins from, well, a rather chaotic mixture of many other proteins in solution. The solution could be anything from a tissue homogenate to a cell lysate, but the goal remains the same: isolation of one specific protein and nothing else – or at least very little else (as in the case of co-immunoprecipitation). There are even protocols adapted from the protein IP procedure for the isolation of chromatin (ChIP) and RNA (RIP), but in this blog post we will only focus on the protein version and provide tips on how to perform better IP experiments.
1. Lysate Preparation
There’s an old adage in the English language: You only get out what you put in, and this saying is true of IP experiments. You want to start with a fair amount of material; aim for between 1 and 3 mg of total protein for every 0.2-0.5 ml of your starting sample volume. You should also aim to keep your target protein as happy as possible throughout the disruptive procedure of cell or tissue lysis. It’s a balancing act really: the ideal lysis buffer will leave proteins in their native conformations and minimize the denaturation of antibody binding sites, but also release enough protein for you to capture and analyse. The Proteintech lab usually uses RIPA buffer, which enables efficient cell lysis and protein solubilization, while avoiding protein degradation and interference with the proteins’ immunoreactivity and biological activity. Using RIPA buffer results in low background in IP and is particularly useful for nuclear membrane disruption for nuclear extracts, but it can denature kinases, so watch out if kinase activity is your goal! And don’t forget to add a protease inhibitor (and a phopshatase inhibitor if you’re looking at phosphorylation!)
You can find our RIPA buffer recipe here. If you’re looking for an alternative to RIPA buffer, our main advice is to go easy on the salt and detergents if you can! Stick to tried and tested buffers (you can find may recipes on scientific forums) and if you do need to tweak your buffer recipes a little, stick within the ranges below recommended by Harlow and Lane, page 231 (see the box below).
2. Choose your antibody wisely
In general, you should consider using a polyclonal antibody, where possible, for the capture of your target protein. Figure 1 provides a good visual explanation as to why this is the case, but in general you’re aiming to establish an antigen-antibody immune complex at the start of the IP procedure. As polyclonals bind multiple epitopes on the target protein, they retain a greater amount of it. As the goal is to wash any unwanted proteins and constituents away later, retention rates need to be pretty high so as not to wash your sample away too! Therefore a good-quality polyclonal should be your first choice for an IP procedure.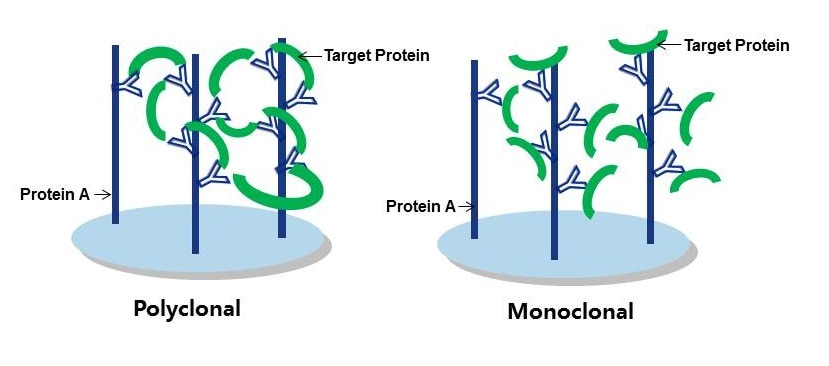
Figure 1: Polyclonal vs Monoclonal as capture antibodies. Polyclonals form tighter binding immune complexes with higher retention rates.
Proteintech’s vast range of polyclonals are ideal for IP as, not only do they fit the job description above, they are also good at recognizing native, intact epitopes. Native epitopes will still retain their three-dimensional confirmations, and as Proteintech antibodies are raised against whole proteins they do the job of recognizing whole, intact proteins in the IP sample rather well. (That being said, it is a good idea to use a monoclonal antibody for detection at the Western blotting (WB) stage, but we’ll get to that later!)
3. To preclear or not to preclear
Preclearing is a step that prevents any non-specific proteins, lipids, carbohydrates or nucleic acids that you don’t want from binding to the solid support you are using in your IP experiment. The step is performed by incubation of the lysate with the solid support (e.g. agarose beads) in the absence of the capture antibody. The solid support used for preclearing, and therefore anything bound to it, is then discarded. This step is dependent on the type of solid support. It also comes down to how good your detection antibody is and how much of your protein of interest might be in the lysate. Some solid supports are more likely to bind non-specifically to certain proteins, so check the manufacturer’s recommendations first , you might be able to skip preclearing all together.
There’s also a preclearing technique that involves the addition of a non-specific antibody of the same isotype as the capture antibody –this can be done to remove anything that might also bind non-specifically to the capture antibody during precipitation.
In general, we do not preclear lysates in the Proteintech lab when performing our IP validations at all for the reason that preclearing is difficult to control when you’re performing a large number of IPs on a regular basis (see also the part about quality of the detection antibody above). It’s not always necessary, and requires careful assessment of the amount of preclearing reagents required to avoid unecessary sample loss.
4. Washing and eluting – mind how you go!
The effect of centrifugation on the outcome of an IP experiment is often underestimated. Centrifugation of the immune complex at too high a speed will disrupt the antibody-antigen interactions in the immune complex and cause sample loss before elution. For that reason we recommend that centrifugation be omitted from the washing and elution steps entirely. Instead, perform washing and obtain elutions using gravitational flow through filter columns loaded in microfuge tubes.
5. Eluted samples vs boiled samples
Traditional measures to improve signal-to-noise ratios aim to retain as much target protein as possible to the detriment of efficient immune complex separation in the final stages of IP. Steps taken toward this end usually involve the omission of reducing agents from the elution buffer or the boiling step prior to SDS-PAGE. Sometimes elution is omitted entirely, and samples still attached to the solid phase are boiled following the washing stages (see Figure 2A). The results are WBs that may have improved target signal, but huge amounts of non-specific staining contributed by the presence of the solid phase (see Figure 2). We strongly recommend that you don’t do this, and elute your samples from the solid support first before gel loading.
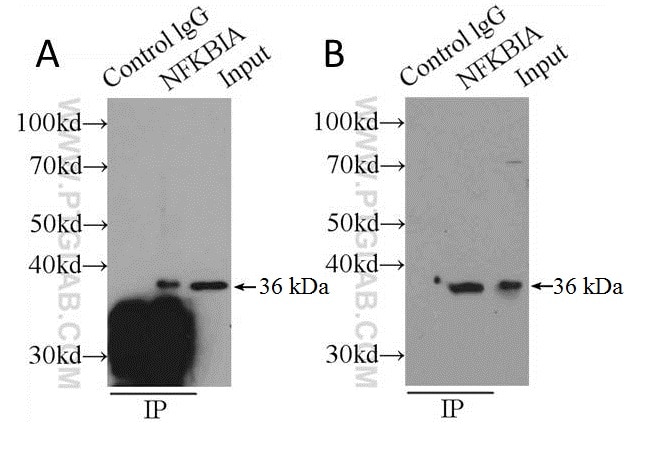
Figure 2: Shows comparison of boiling samples (still attached to solid support) immediately after washing (A) and after elution (B) using PH 2 buffer (150 mM glycine, 500 mM NaCl) and neutralized before loading on an SDS-PAGE gel. Capture and detection antibody was anti-NFKBIA (cat. no. 10268-1-P) secondary detection was done using HRP-conjugated Protein A (see the section on alternative detection methods). Cell lysate: HeLa.
6. Keep your fractions!
It’s advisable to hang on to all of your IP fractions (including samples from any preclearing you perform) to anaylze by SDS-PAGE later, should your initial attempts at immunopreciptation be unsuccessful.
7. Antibody pairs
The use of antibody pairs, i.e. a capture antibody from one species, and an antibody for WB detection from another, is an additional factor to consider when planning an IP experiment. The antibody selection process involved should ensure both antibodies recognize different epitopes of the target protein, in addition to originating from different species. The antibody type (i.e. polyclonal or monoclonal) is also important. We suggest a combination of a polyclonal capture antibody and a monoclonal antibody for detection where possible; this ensures maximum capture efficiency with high detection specificity. See the example in Figure 3.
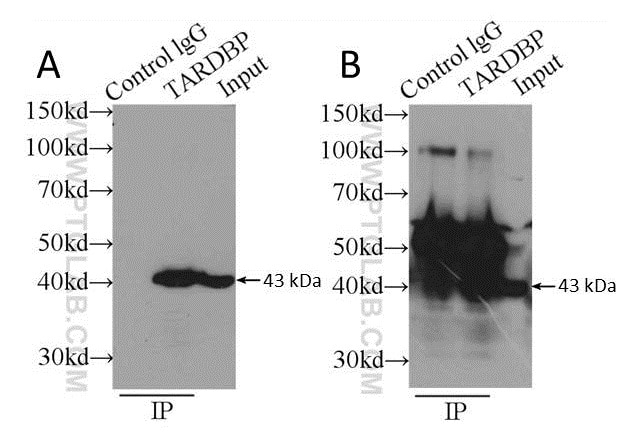
Figure 3: Comparison of using a polyclonal capture antibody (10782-2-AP) and a monoclonal detection antibody (60019-1-Ig) (A) with the same antibody (10782-2-AP) for both capture and detection (B). Secondary detection was performed using HRP-goat anti-mouse (A) and HRP-goat anti-rabbit (B). Cell lysate used was K-562 whole cell lysate.
8. Try an alternative detection method!
Anyone who’s ever done an IP experiment before will know that the heavy chain (HC) and light chain (LC) of the capture antibody can interfere with your target signal. This is an unavoidable artifact of having to use the same antibody for capture and detection, and then use a species specific antibody on top of that for secondary detection.
References
Harlow, Ed, and David Lane. Using Antibodies. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 1999.
Bonifacino, Juan S. et al. Current Protocols in Immunology 8.3.1-8.3.28, New York: John Wiley, 2001.
Related Content
Choosing the right antibody format for immunoprecipitation
How to obtain low background in immunoprecipitation
Immunoprecipitation without additional bands
Immunopreciptation of low abundant proteins
What GFP-Trap should I use for my immunoprecipitation?
Mass spec-compatible immunoprecipiation for GFP, mNeonGreen, Myc, RFP, Spot, and TurboGFP

Support
Newsletter Signup
Stay up-to-date with our latest news and events. New to Proteintech? Get 10% off your first order when you sign up.
Able™