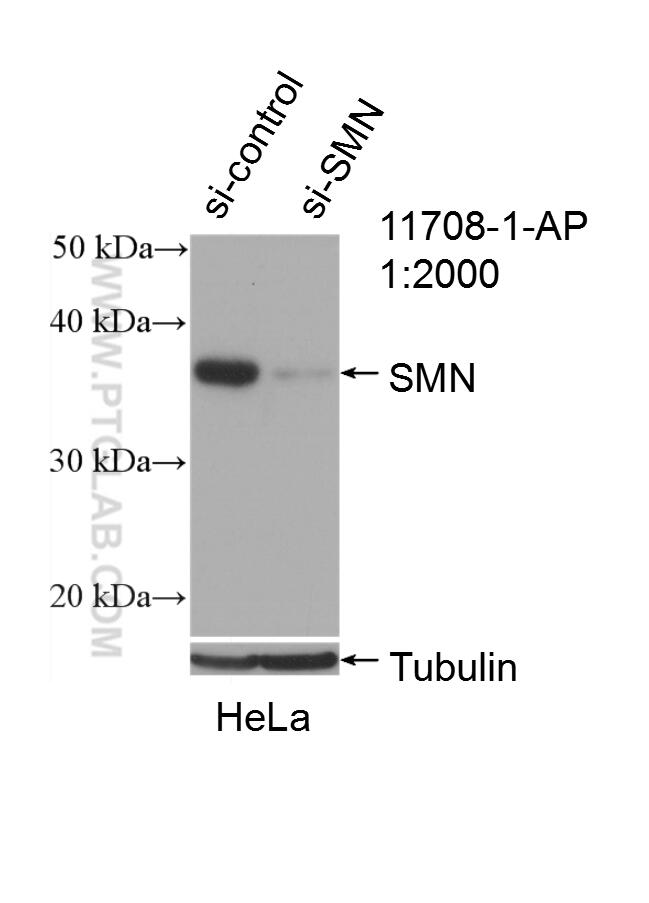
analysis of HeLa cells using SMN Polyclonal antibody (11708-1-AP)")
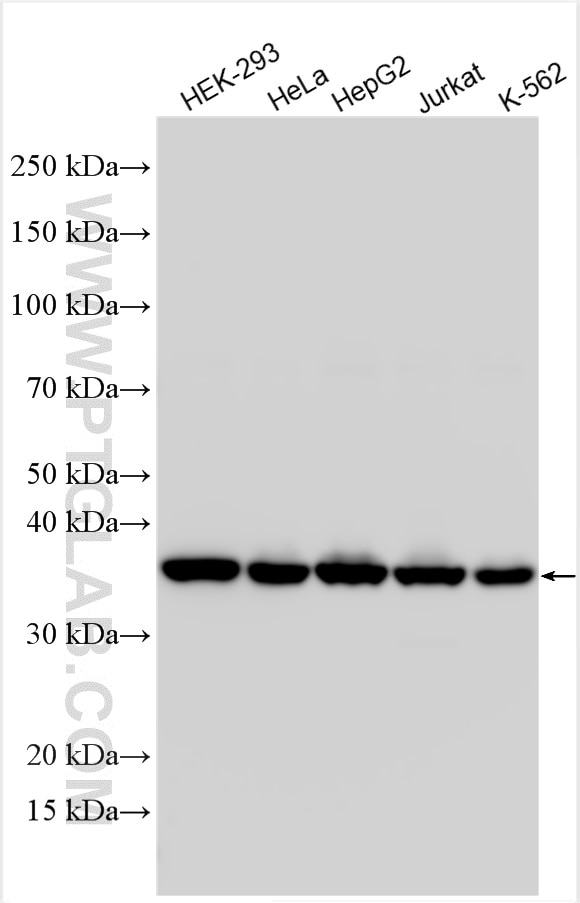
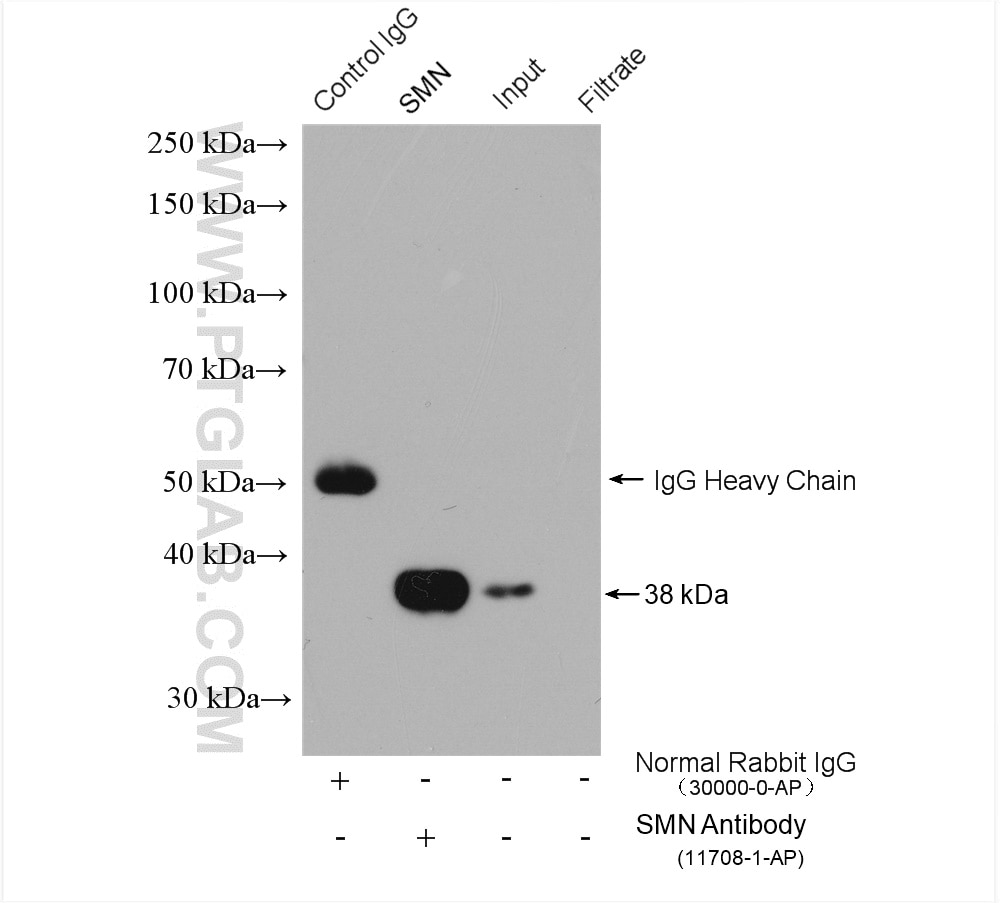
 analysis of various lysates using SMN Polyclonal antibody (11708-1-AP)")
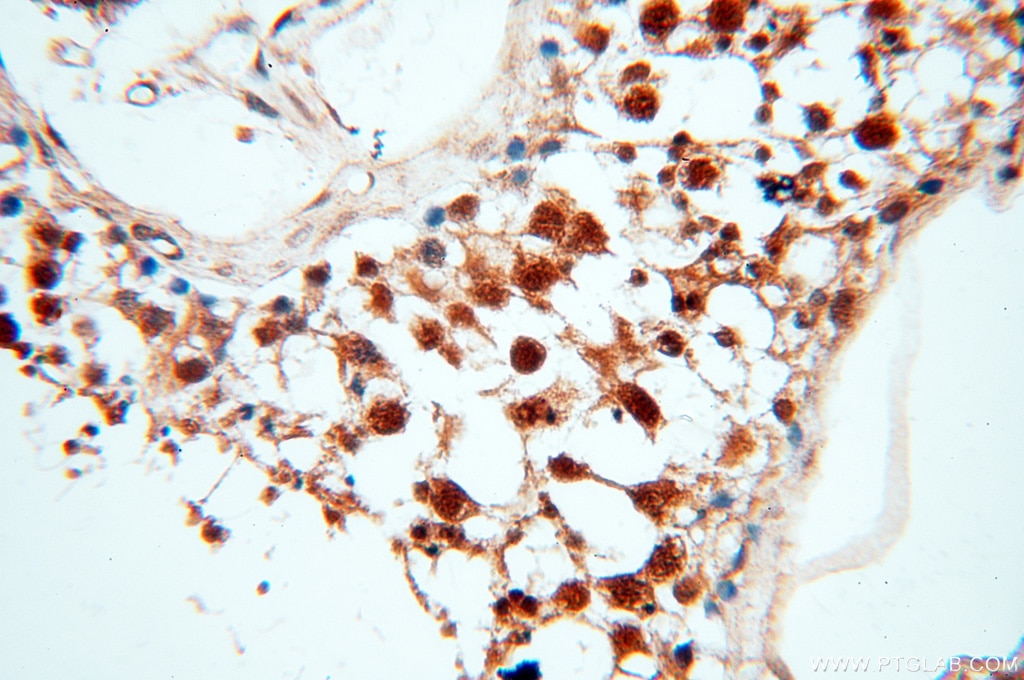
 analysis of mouse testis tissue using SMN Polyclonal antibody (11708-1-AP)")
 experiment of HEK-293 cells using SMN Polyclonal antibody (11708-1-AP)")
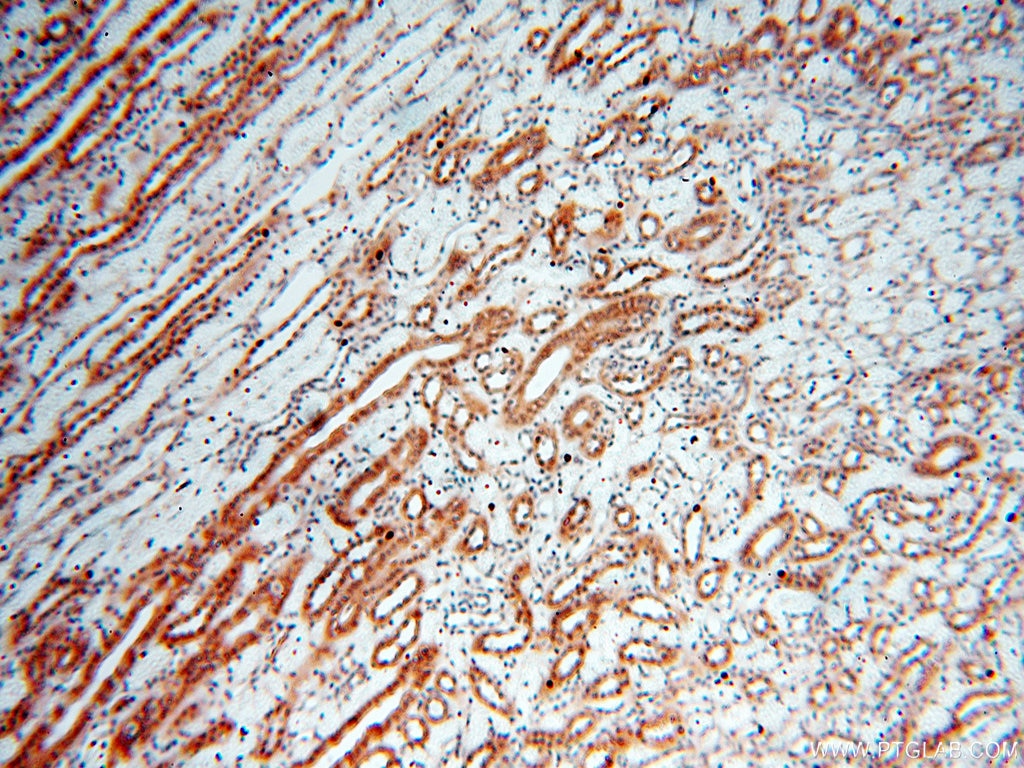
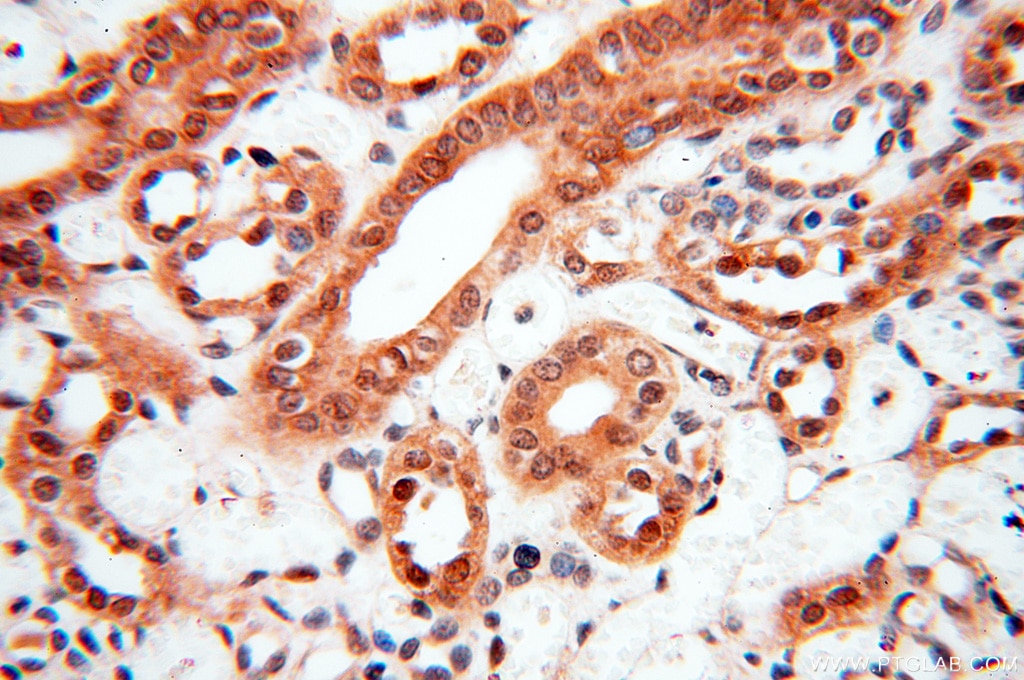
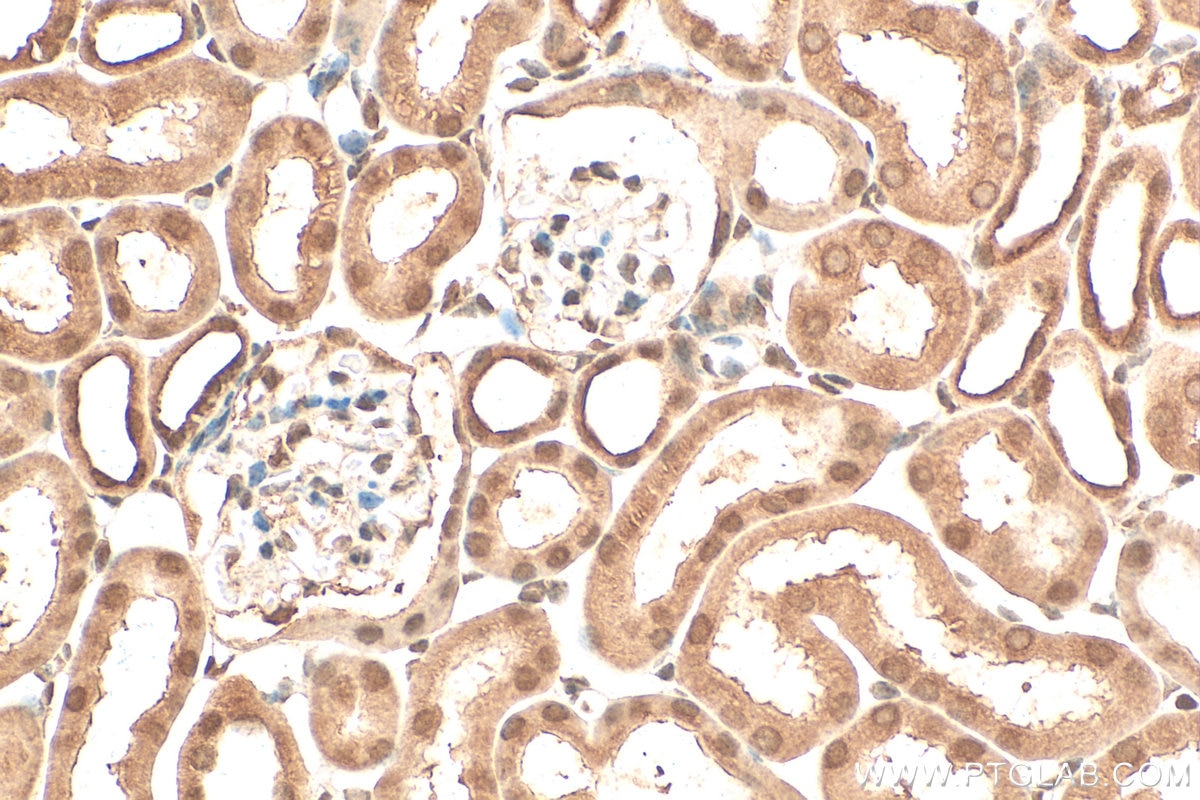
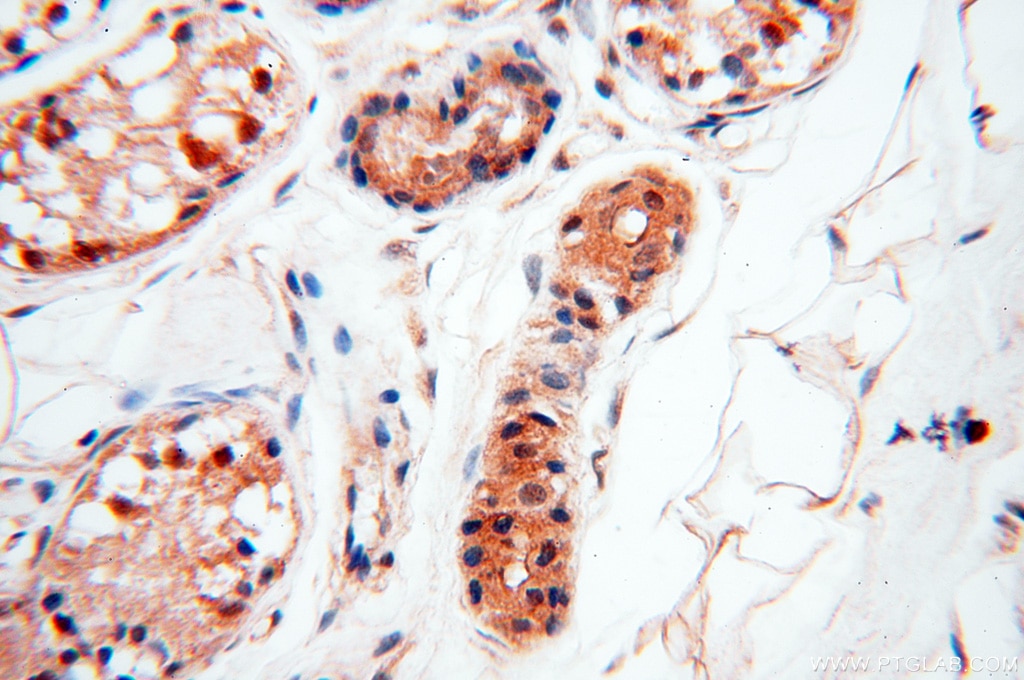
 staining of human kidney tissue using SMN Polyclonal antibody (11708-1-AP)")
 staining of human kidney tissue using SMN Polyclonal antibody (11708-1-AP)")
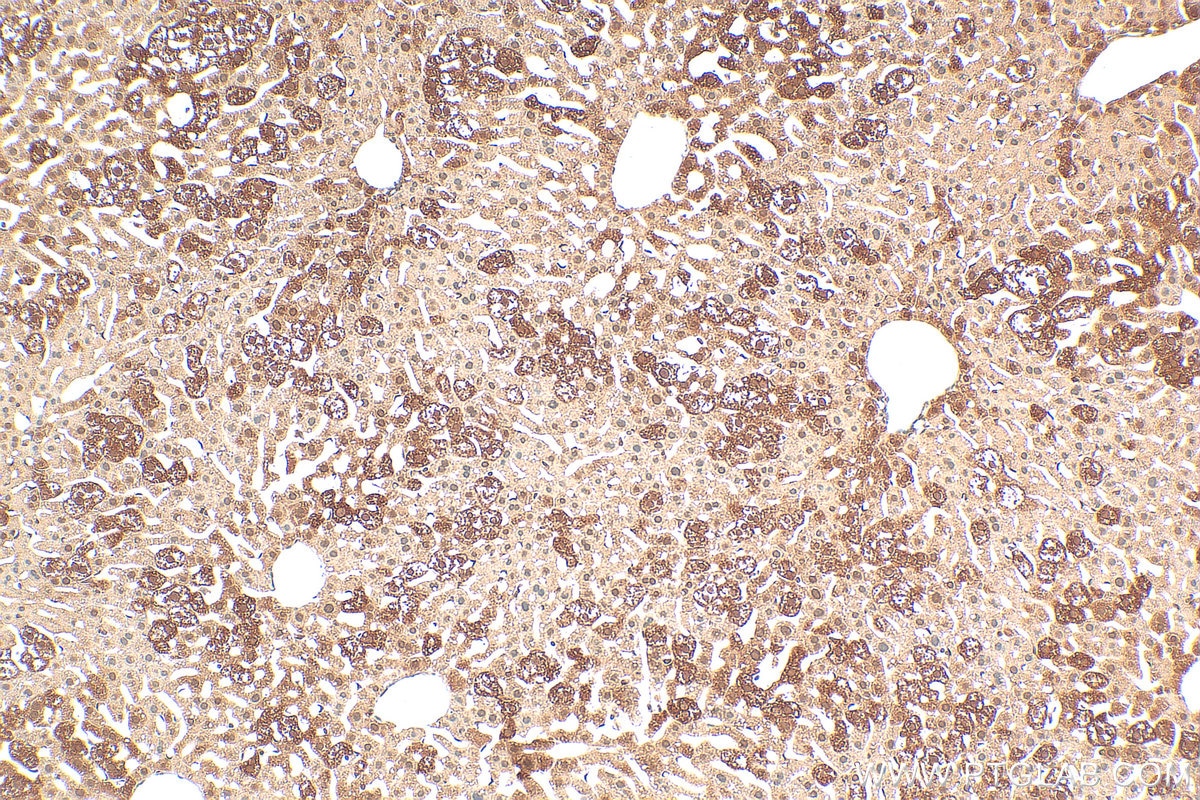
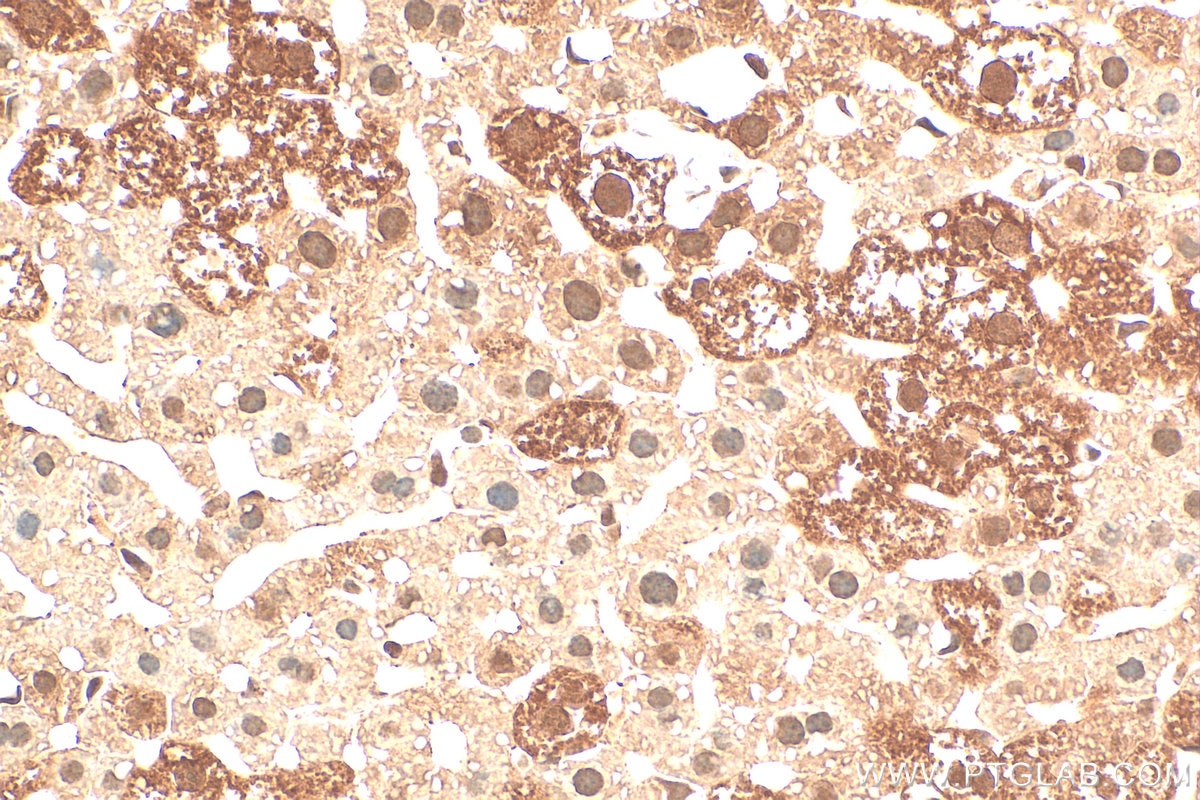
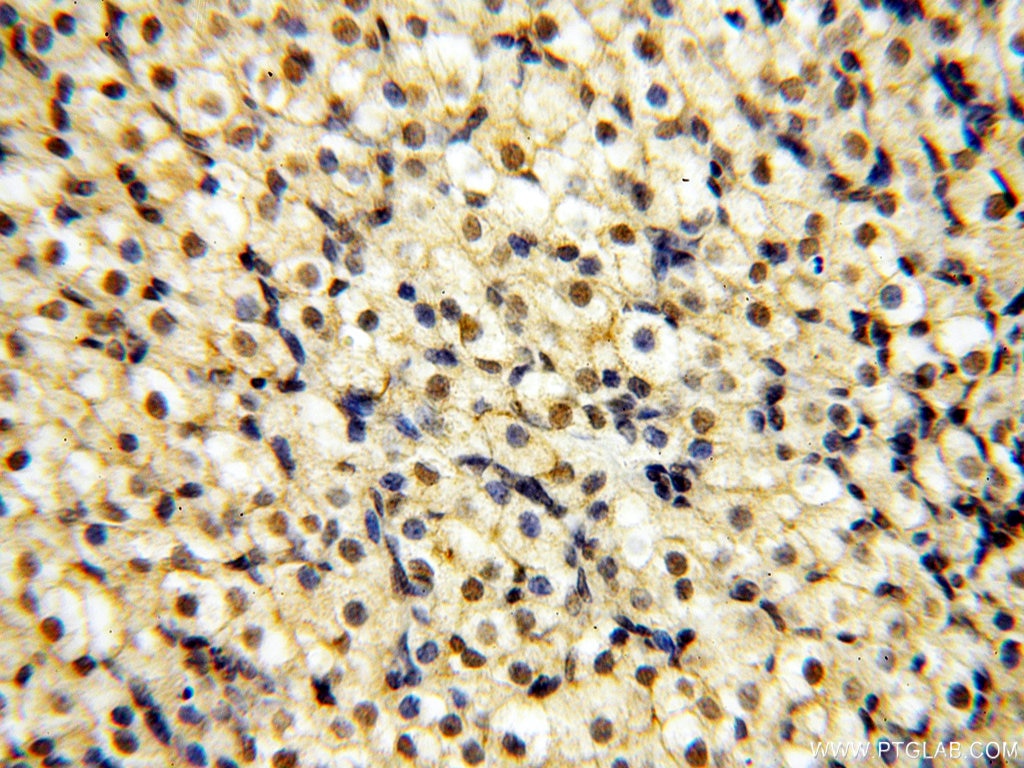
 staining of mouse liver tissue using SMN Polyclonal antibody (11708-1-AP)")
 staining of mouse liver tissue using SMN Polyclonal antibody (11708-1-AP)")
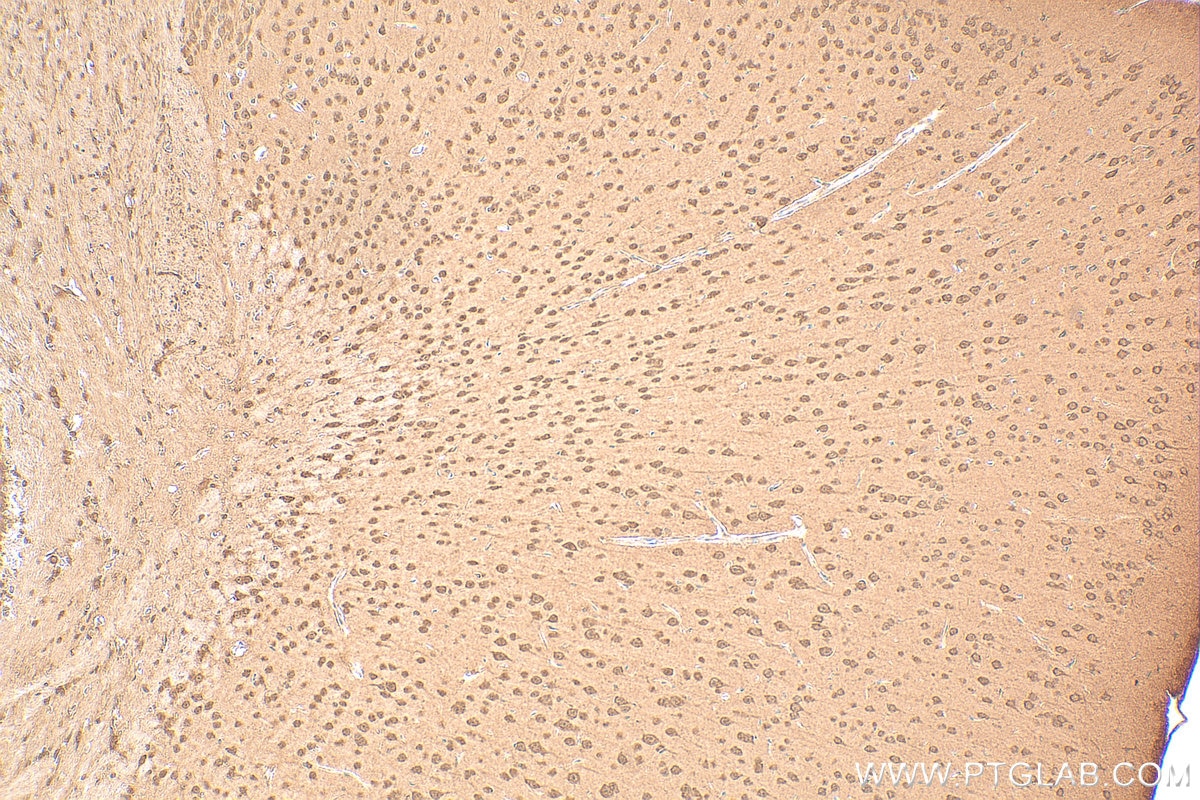
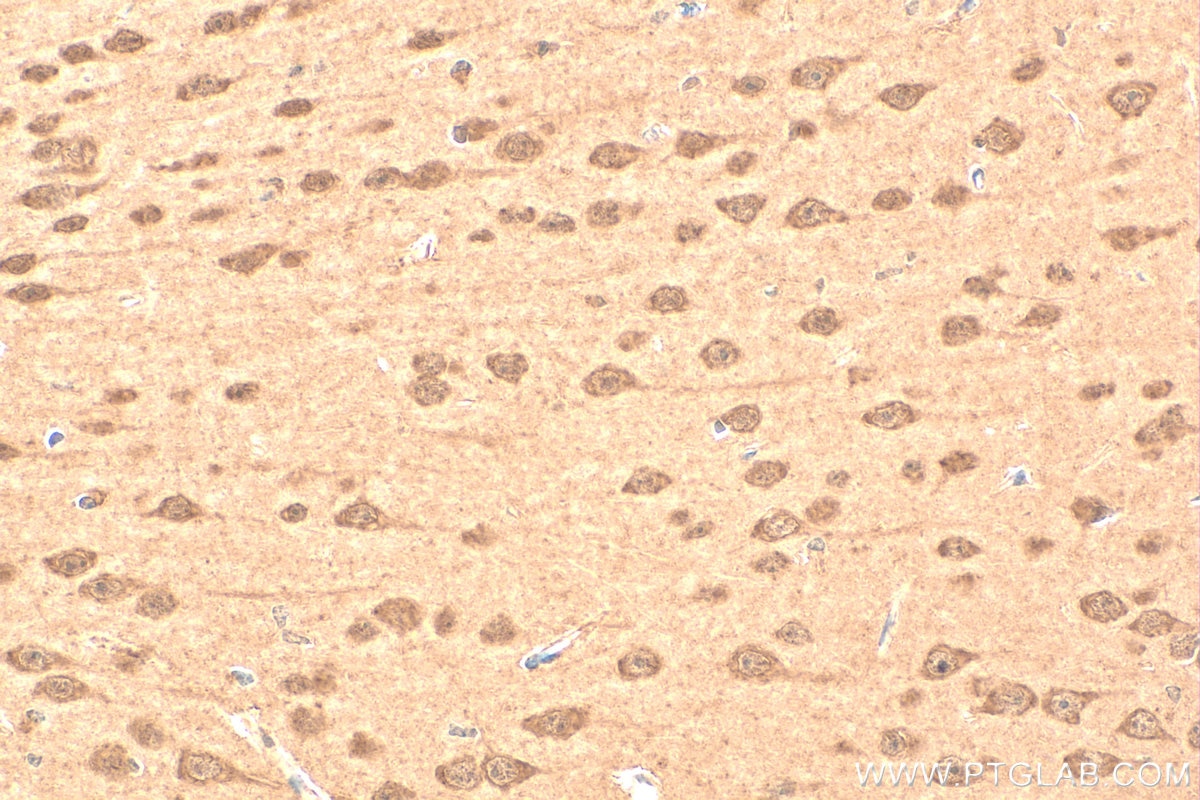
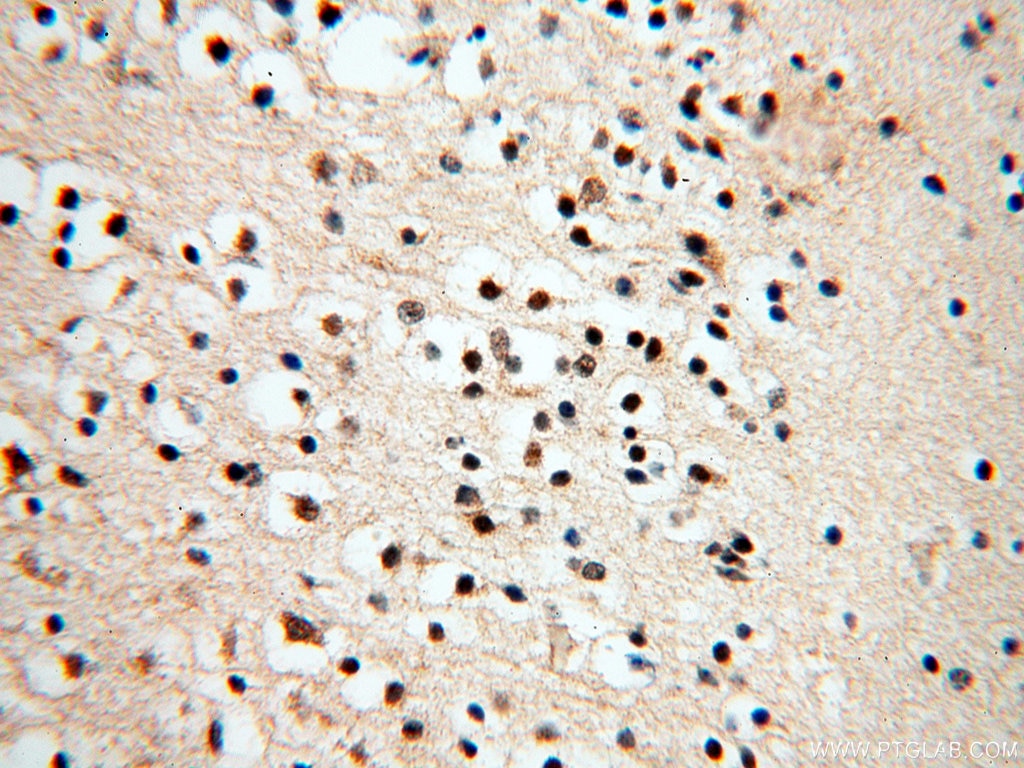
 staining of mouse brain tissue using SMN Polyclonal antibody (11708-1-AP)")
 staining of mouse brain tissue using SMN Polyclonal antibody (11708-1-AP)")
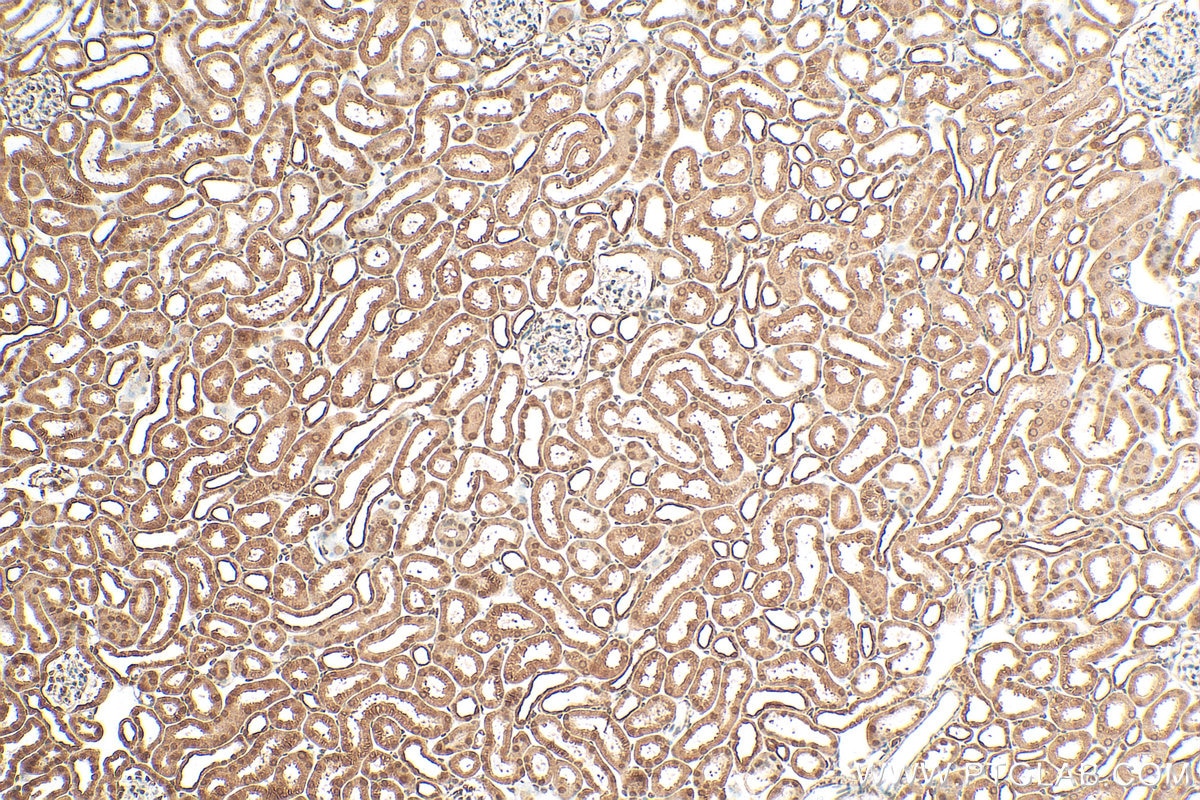
 staining of mouse kidney tissue using SMN Polyclonal antibody (11708-1-AP)")
 staining of mouse kidney tissue using SMN Polyclonal antibody (11708-1-AP)")
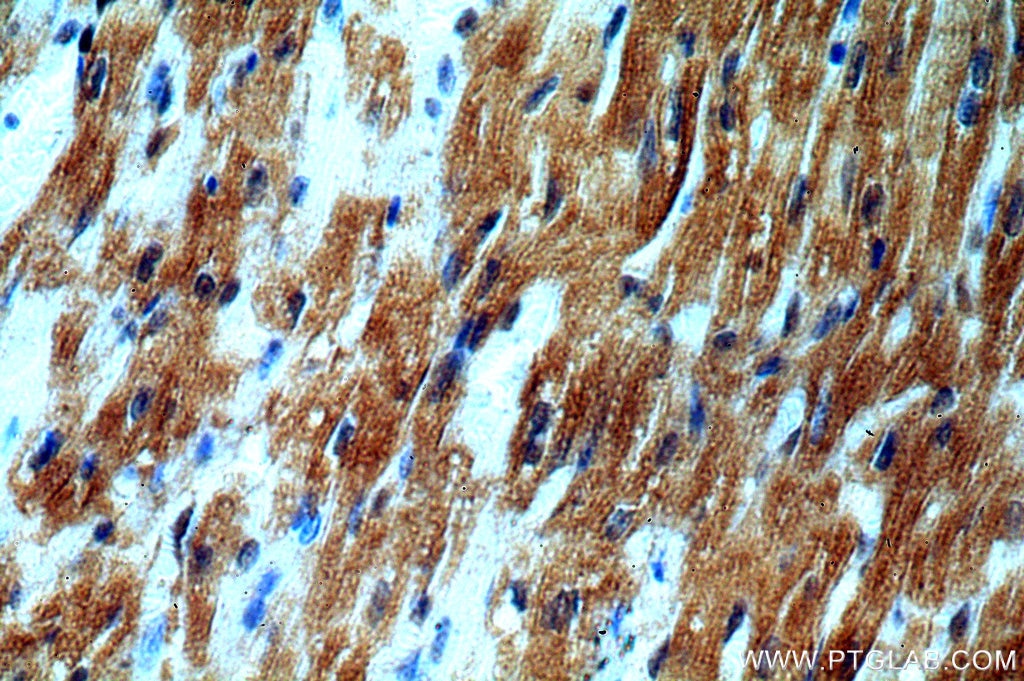
 staining of human heart tissue using SMN Polyclonal antibody (11708-1-AP)")
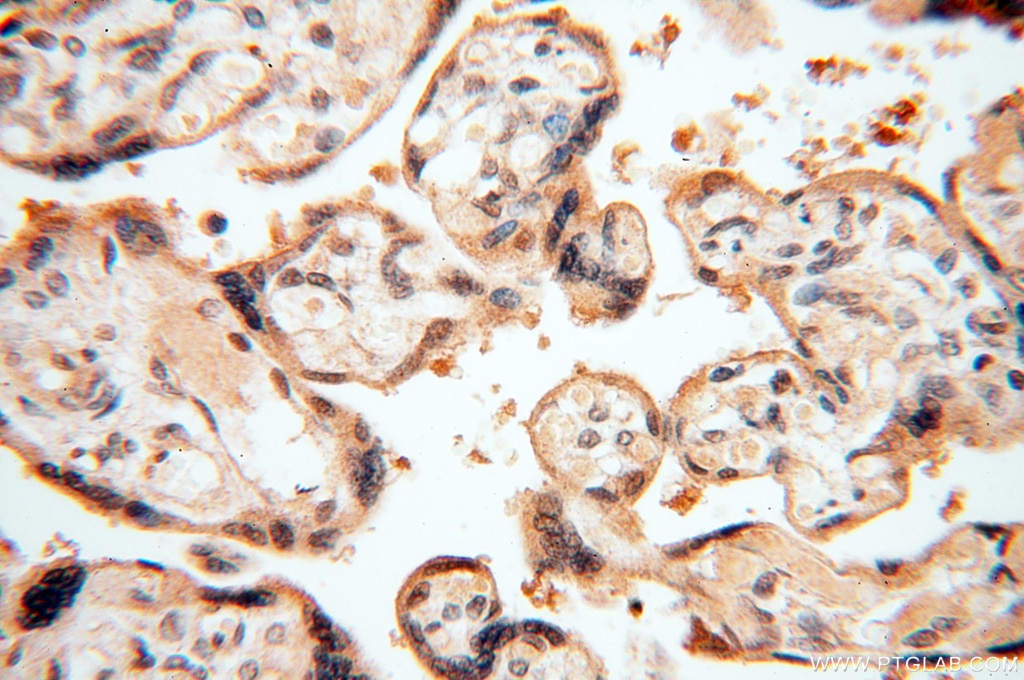
 staining of human placenta tissue using SMN Polyclonal antibody (11708-1-AP)")
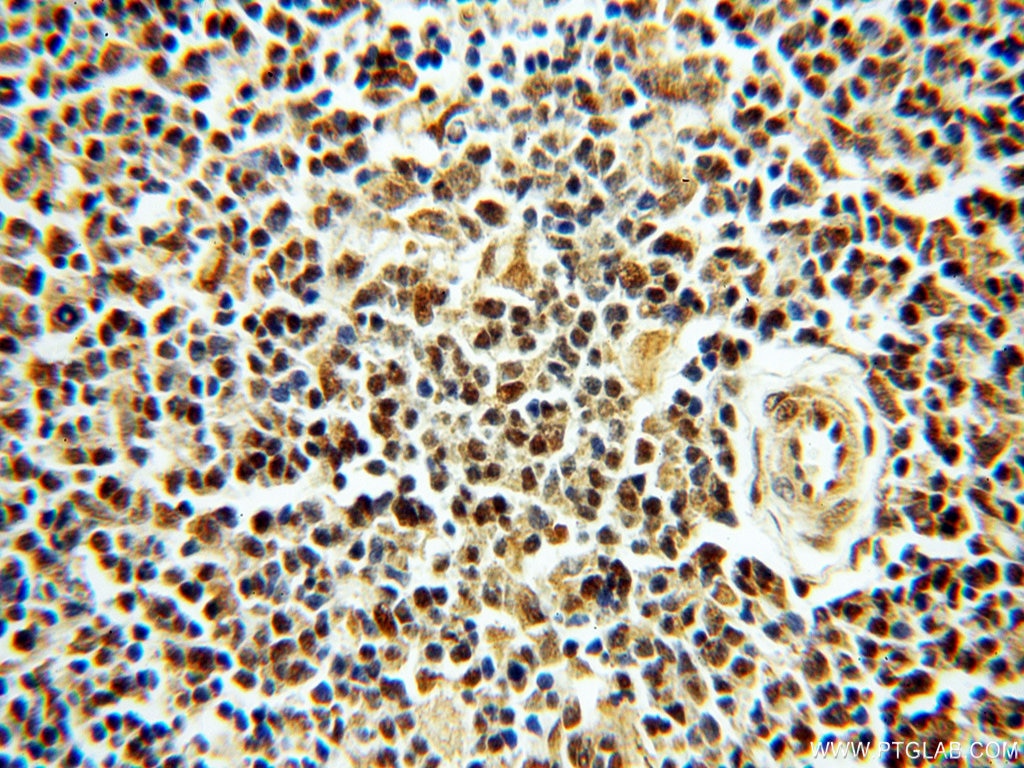
 staining of human testis tissue using SMN Polyclonal antibody (11708-1-AP)")
 staining of human skin tissue using SMN Polyclonal antibody (11708-1-AP)")
 staining of human brain tissue using SMN Polyclonal antibody (11708-1-AP)")
 staining of human spleen tissue using SMN Polyclonal antibody (11708-1-AP)")
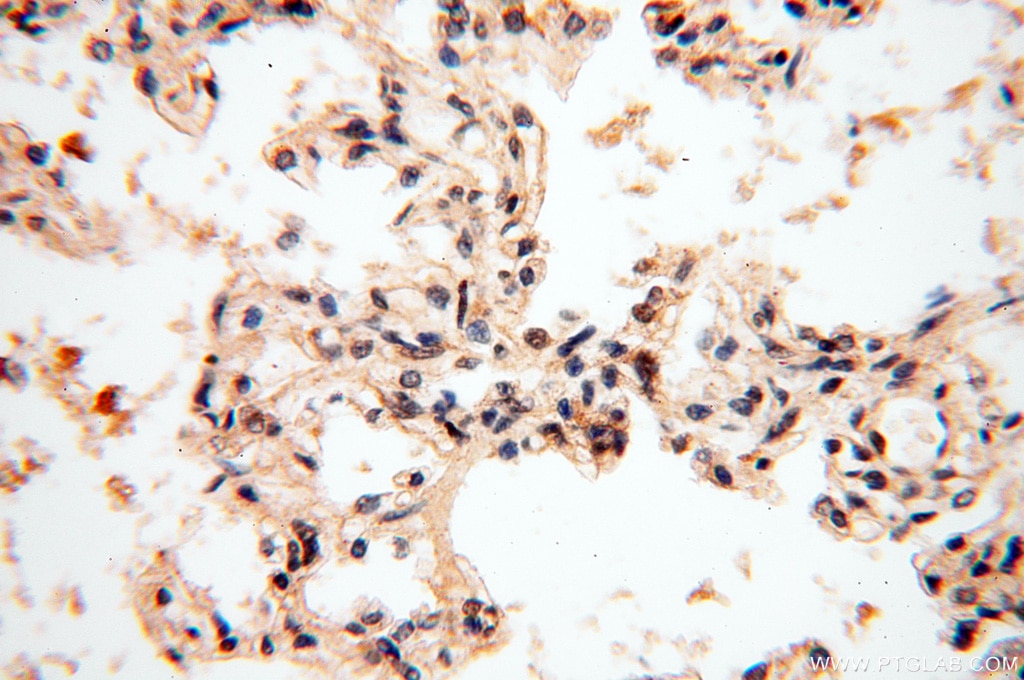
 staining of human lung tissue using SMN Polyclonal antibody (11708-1-AP)")
 staining of human ovary tissue using SMN Polyclonal antibody (11708-1-AP)")
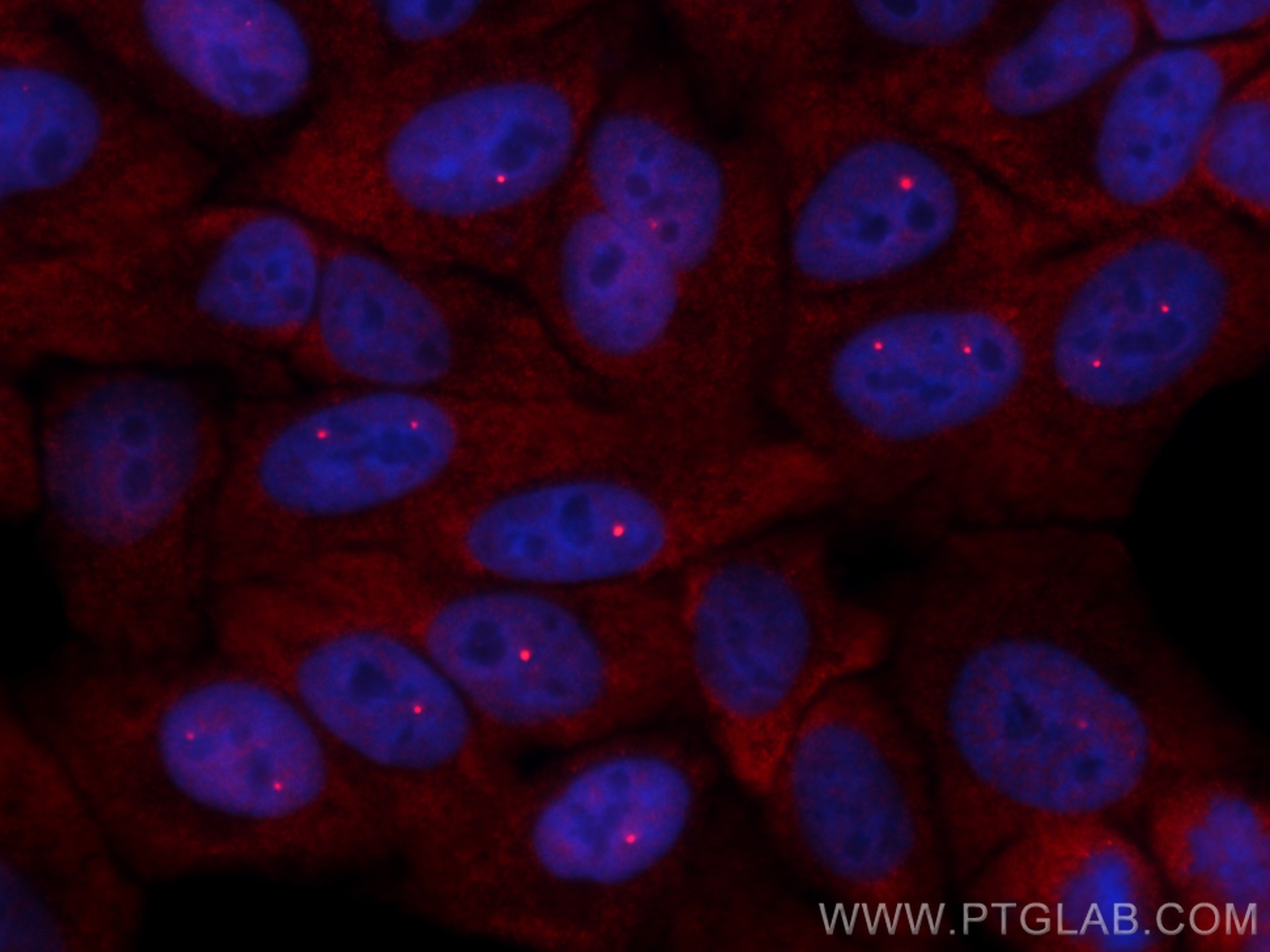
 / fluorescent staining of HepG2 cells using SMN Polyclonal antibody (11708-1-AP)")
Tested Applications
| Positive WB detected in | HEK-293 cells, HeLa cells, mouse testis tissue, HepG2 cells, Jurkat cells, K-562 cells |
| Positive IP detected in | HEK-293 cells |
| Positive IHC detected in | human kidney tissue, human brain tissue, human heart tissue, human lung tissue, human ovary tissue, human placenta tissue, human skin tissue, human spleen tissue, human testis tissue, mouse brain tissue, mouse kidney tissue, mouse liver tissue Note: suggested antigen retrieval with TE buffer pH 9.0; (*) Alternatively, antigen retrieval may be performed with citrate buffer pH 6.0 |
| Positive IF/ICC detected in | HepG2 cells |
Recommended dilution
| Application | Dilution |
|---|---|
| Western Blot (WB) | WB : 1:2000-1:16000 |
| Immunoprecipitation (IP) | IP : 0.5-4.0 ug for 1.0-3.0 mg of total protein lysate |
| Immunohistochemistry (IHC) | IHC : 1:50-1:200 |
| Immunofluorescence (IF)/ICC | IF/ICC : 1:750-1:3000 |
| It is recommended that this reagent should be titrated in each testing system to obtain optimal results. | |
| Sample-dependent, Check data in validation data gallery. | |
Published Applications
| KD/KO | See 1 publications below |
| WB | See 4 publications below |
| IF | See 3 publications below |
| IP | See 1 publications below |
| ELISA | See 4 publications below |
Product Information
11708-1-AP targets SMN in WB, IHC, IF/ICC, IP, ELISA applications and shows reactivity with human, mouse, rat samples.
| Tested Reactivity | human, mouse, rat |
| Cited Reactivity | human, mouse, rat |
| Host / Isotype | Rabbit / IgG |
| Class | Polyclonal |
| Type | Antibody |
| Immunogen |
CatNo: Ag2260 Product name: Recombinant human SMN2 protein Source: e coli.-derived, PGEX-4T Tag: GST Domain: 1-282 aa of BC000908 Sequence: MAMSSGGSGGGVPEQEDSVLFRRGTGQSDDSDIWDDTALIKAYDKAVASFKHALKNGDICETSGKPKTTPKRKPAKKNKSQKKNTAASLQQWKVGDKCSAIWSEDGCIYPATIASIDFKRETCVVVYTGYGNREEQNLSDLLSPICEVANNIEQNAQENENESQVSTDESENSRSPGNKSDNIKPKSAPWNSFLPPPPPMPGPRLGPGKPGLKFNGPPPPPPPPPPHLLSCWLPPFPSGPPIIPPPPPICPDSLDDADALGSMLISWYMSGYHTGYYMEMLA Predict reactive species |
| Full Name | survival of motor neuron 2, centromeric |
| Calculated Molecular Weight | 282 aa, 30 kDa |
| Observed Molecular Weight | 38 kDa |
| GenBank Accession Number | BC000908 |
| Gene Symbol | SMN |
| Gene ID (NCBI) | 6607 |
| RRID | AB_2255114 |
| Conjugate | Unconjugated |
| Form | Liquid |
| Purification Method | Antigen affinity purification |
| UNIPROT ID | Q16637 |
| Storage Buffer | PBS with 0.02% sodium azide and 50% glycerol, pH 7.3. |
| Storage Conditions | Store at -20°C. Stable for one year after shipment. Aliquoting is unnecessary for -20oC storage. 20ul sizes contain 0.1% BSA. |
Background Information
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease characterized by loss of anterior horn cells in the spinal cord and concomitant symmetrical muscle weakness and atrophy (PMID: 16364894 ). SMA is caused by deletion or mutations of the survival motor neuron (SMN1) gene. SMA patients lack a functional SMN1 gene, but they possess an intact SMN2 gene, which though nearly identical to SMN1, is only partially functional (PMID: 17355180). A large majority of SMN2 transcripts lack exon 7, resulting in production of a truncated, less stable SMN protein (PMID: 10369862). The level of SMN protein correlates with phenotypic severity of SMA. This antibody, 11708-1-AP, raised against the recombinant full-length human SMN2 protein, recognizes all isoforms of SMN protein.
Protocols
| Product Specific Protocols | |
|---|---|
| IF protocol for SMN antibody 11708-1-AP | Download protocol |
| IHC protocol for SMN antibody 11708-1-AP | Download protocol |
| IP protocol for SMN antibody 11708-1-AP | Download protocol |
| WB protocol for SMN antibody 11708-1-AP | Download protocol |
| Standard Protocols | |
|---|---|
| Click here to view our Standard Protocols |
Publications
| Species | Application | Title |
|---|---|---|
Nat Commun SNUPN deficiency causes a recessive muscular dystrophy due to RNA mis-splicing and ECM dysregulation | ||
Dev Cell DDX20 is required for cell-cycle reentry of prospermatogonia and establishment of spermatogonial stem cell pool during testicular development in mice | ||
Hum Mol Genet Low levels of Survival Motor Neuron protein are sufficient for normal muscle function in the SMNΔ7 mouse model of SMA. | ||
Hum Mol Genet SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNΔ7 mouse model of SMA. | ||
Neurobiol Dis Dual SMN inducing therapies can rescue survival and motor unit function in symptomatic ∆7SMA mice. | ||
Hum Mol Genet Intragenic complementation of amino and carboxy terminal SMN missense mutations can rescue Smn null mice.
|
Reviews
The reviews below have been submitted by verified Proteintech customers who received an incentive for providing their feedback.
FH jummoin (Verified Customer) (12-11-2025) | Good SMN antibody with clear, specific bands and reliable performance in WB and IF. Useful for studying SMN expression across different models.
|
FH randi (Verified Customer) (12-11-2025) | Reliable SMN antibody that detects all SMN isoforms and works well in Western blot, IHC, IF, and IP with clear, specific results across human and rodent samples. Suitable for studies involving SMN protein expression in cellular and tissue analyses.
|
FH Rachel (Verified Customer) (08-19-2019) | Worked well in MSD immunoassays and cell based assays.
|