
Mitochondria: More than just the powerhouse of the cell
An introduction to mitochondria in health and disease.
By Sophie Quick
Mitochondria are the result of a billion-year old fusion of primitive bacteria and early eukaryotic cells, resulting in a species-defining endosymbiosis. These organelles not only generate the cell’s chemical energy through ATP-proton pumps but have emerged as key players in a number of cellular processes including signalling, differentiation and apoptosis. As many as thousands or as few as tens of mitochondria may be found within a human cell, comprising a highly dynamic network constantly undergoing fission and fusion.
Inherited or spontaneous mutations in the mitochondrial DNA (mtDNA) result in a specific subset of disorders known as mitochondrial diseases. In addition, mitochondria have been implicated in many common human diseases. Alterations to key effector proteins that influence cellular processes and mitochondrial dynamics have been identified in neurodegeneration, cardiovascular diseases and cancer, to name a few. Here we will explore research published in the last 18 months showing the role that some of these proteins may play in the pathology or even the treatment of selected examples of these diseases.
Mitochondria in Parkinson’s disease
Mitochondria have long been linked to Parkinson’s disease (PD), a degenerative condition affecting dopamine-producing brain cells in the substantia nigra. These dopaminergic neurons progressively die, through a process involving the loss of function in the mitochondria and a build-up proteinaceous deposits mainly consisting of α-synuclein. The process is not fully understood, but leads to characteristic motor impairment symptoms which current treatments can only relieve and not cure.
Mutations in parkin, an E3 ligase involved in the degradation of damaged proteins and mitochondria, were first identified as a cause of PD almost 20 years ago. These mutations lead to an accumulation of parkin substrates and have historically been linked to a rare familial form of the disease. However, a recent study (Andersen et al, 2016) has indicated that oxidative stress affects parkin in the same way. As oxidative stress is one of the main factors for sporadic PD, the implications for parkin now extends to 95% of PD cases. Using a mouse model of PD, the researchers also investigated the signalling pathway involved in parkin dysfunction and found two key proteins that would be suitable targets for drug development. TFEB is involved in mitochondrial degradation while PGC-1α is involved in mitochondrial synthesis and both were seen to be down-regulated in oxidative stress. Interestingly, increasing expression of PGC-1α in the PD mouse model resulted in a recovery of mitochondrial function and a prevention of neuron death. This indicates a potential target for a preventative treatment for the disease.
Research published in June (Di Maio, 2016) has highlighted how the two hallmarks of pathogenesis – the build-up of α-synuclein and mitochondrial dysfunction – are linked through the particular mitochondrial receptor TOM20 (Translocase of the Outer Machinery 20). TOM20 is found on the outer membrane as a part of the mitochondrial protein import machinery. The study showed in a rodent model of PD that types of α-synuclein bind with strong affinity to TOM20, preventing interaction with the co-receptor TOM22 and affecting the energy producing function of the mitochondria. The inefficiency of affected mitochondria combined with the resulting toxicity of accumulated α-synuclein was confirmed in brain tissue from PD patients, and the team even showed in cell cultures that preventing α-synuclein from binding to TOM20 was enough to prevent its damage. The colocalization of mitochondria with α-synuclein through TOM20 provides an interesting target in PD research, though future work is required to confirm whether altering this protein could improve disease symptoms in vivo.
Related Products
| TOM20 | TOM22 |
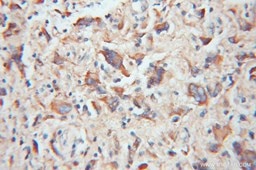 |
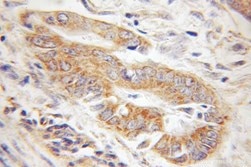 |
| IHC of paraffin-embedded human gliomas using 11802-1-AP(TOM20 antibody) at dilution of 1:100 (under 10x lens) | IHC of paraffin-embedded human colon cancer using 11278-1-AP(TOMM22 antibody) at dilution of 1:100 (under 10x lens) |
Mitochondria in Cancers
The uncontrolled division of tumour cells in cancer is accompanied by a switch in the way cells produce energy, known as the Warburg effect, from mitochondrial oxidative phosphorylation to glycolysis. While the original theory that mitochondrial dysfunction is the root cause of cancer has been replaced with the understanding of mutated oncogenes, it is clear that mitochondria remain key in tumour development, affecting pathways such as apoptotic signalling and redox regulation. Important insight into cancer progression emerged last year when a group (Kashatus et al, 2015) demonstrated how cancer cells in fact hijack mitochondria to support enhanced anabolic respiration and promote tumour growth. Mutations in the Ras gene, which are found in a number of all cancers, cause mitochondria to undergo fission at an abnormally high rate both in cell cultures and in rodent models. Furthermore, by knocking down the mitochondrial replication, researchers were able to block tumour growth.
Mitochondrial mediated apoptosis has also become a point of interest as a potential treatment for cancer cells. By targeting particular mitochondrial proteins researchers proposed they would be able to initiate cell death through the permeabilisation of the mitochondrial membrane, leaking proteins such as cytochrome c which activate destructive caspase pathways. Indeed, in May this year a Nature paper reported how researchers (Iyer et al, 2016) were able to directly activate Bak with a targeted antibody to trigger apoptosis. They were able to identify the three-dimensional structure of the activation site which may inform the design and development of drugs with a novel target site.
Mitochondria in Cardiovascular diseases
Coronary heart disease (CHD), the leading cause of death worldwide, is described as the lack of oxygen supplied to cardiomyocytes. Hypoxia curbs ATP production, causing cellular stress and an overproduction of reactive oxygen species (ROS) by mitochondria. The accumulation of ROS leads to apoptosis and necrosis of cardiac cells so has been closely linked by researchers to disease pathogenesis.
Defective mitochondrial respiratory complexes are key players in the mechanism of this disease progression. The ability of healthy mitochondria to respond to hypoxia and increase capacity, tapping into a ‘reserve’ of ATP production, was proposed to be regulated by a factor which may be damaged in CHD. Last year, a team (Pflenger et al, 2015) provided evidence that Sdh of complex II is the metabolic sensor that controls cell survival after hypoxia. Targeting this protein could provide new information regarding the progression of this disease.
The dynamic nature of mitochondria also plays an important role in disease progression. The network of fission and fusion is regulated by a number of proteins including mitochondrial fusion proteins Mfn1 and Mfn2 (Mitofusin 1 and 2). Overexpression of these proteins seems to protect against the increased production of ROS after the cell undergoes hypoxia. A team at the Max Planck Institute last year (Mourier et al, 2015) showed that Mfn2-deficient cardiac cells had impaired metabolism, which they were able to partially rescue by supplementing the cells with an enzyme generated further down the pathway, indicating a potential therapeutic option for patients with Mfn2 mutations.
Through the production of ROS via a number of mechanisms, mitochondria influence the progression of heart disease. As research progresses, they are becoming interesting targets for cardioprotection.
| MFN1 |
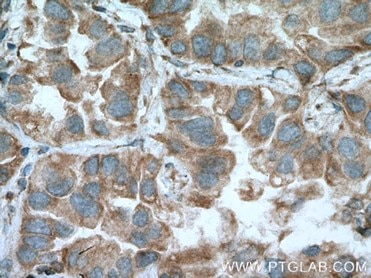 |
| IHC of paraffin-embedded human lung cancer tissue slide using 13798-1-AP( MFN1 Antibody) at dilution of 1:200 (under 40x lens). |
Future Perspectives
Many of the proteins mentioned here can be linked to a range of diseases based on their effects on mitochondrial dynamics such as fusion, fission and mitophagy and so provide an interesting target for a range of research areas. One school of thought pioneered by mitochondrial researchers at the Children’s Hospital of Philadelphia suggests that treating disease by concentrating on the organ with symptoms overlooks bioenergetics entirely (Picard et al, 2015). This perspective proposes the idea that the interaction between the essential energy genes encoded by the mtDNA and those within the nuclear DNA play a more important part in health and disease. Though an organ-focused approach is currently favoured when treating disease, perhaps a mitochondrial-focused approach could help to inspire new research and the development of new treatments. Meanwhile, the rapidly advancing area of mitochondrial replacement therapy in embryos, where faulty mtDNa of a mother with mitochondrial disease is replaced with healthy mtDNA, could pave the way for truly personalised medicine. This novel area requires researchers to consider a number of issues (Falk et al, 2016) as altering the inheritance of genetic material has both physical and ethical implications. Understanding the role of the mitochondria at all stages of development will develop our ability to model and to treat a wide range of diseases.
References
1. Andersen, Julie et al. Detrimental effects of oxidative losses in parkin activity in a model of sporadic Parkinson’s disease are attenuated by restoration of PGC1 alpha. Neurobiology of Disease, June 2016
2. Di Maio, Roberto et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease, Science Translational Medicine 08 Jun 2016: Vol. 8, Issue 342, pp. 342ra78
3. Kashatus, Jennifer et al. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Molecular Cell, Volume 57, Issue 3
4. Iyer, Sweta et al. Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies. Nature Communications, 2016; 7: 11734
5. Pfleger, J et al, Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival, Cell Death and Disease (2015) 6, e1835;
6. Mourier, A. et al, Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. The Journal of Cell Biology, 2015; 208 (4): 429
7. Picard, M. et al. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proceedings of the National Academy of Sciences, 2015
8. Falk, Marni J. et al. Mitochondrial Replacement Techniques — Implications for the Clinical Community. New England Journal of Medicine, 2016
Able™