
Cell culture protocol
Our protocol for subculturing and counting cells, and creating cell banks.
| Cell Culture Protocol | |
| Standard passaging (subculture) | Cell banks (cryopreservation) |
| Adherent cells | Preparing cell banks |
| Suspension cells | Thawing cells from banks |
| Cell counting | Cell transfection |
Standard passaging (subculture)
Passaging refers to the diluting of cells that have reached high confluence to supplement cells with fresh medium to enable continuous culture propagation.
For mammalian cells, passaging should be performed when cells are toward the end of the logarithmic growth phase, so before they reach the stationary phase (Figure 3). For adherent cells that usually means reaching 80–90% confluence (there is still space for growth on a culture dish); for suspension culture, it is when cells start to clump and the culture becomes cloudy, especially if a dish is shaken slightly.
Passaging of cells at the stationary phase is not recommended because they tend to take longer to begin the logarithmic growth phase upon seeding. Additionally, the build-up of lactic acid in dense cultures may impact cell metabolism.
Adherent cells
-
Remove medium.
-
Wash cells with calcium- and magnesium-free balanced salt solution (phosphate buffered saline, Hanks’ balanced salt solution, etc.
-
Add detaching agent (e.g., trypsin). Incubate at 37°C until cells are fully detached from the dish (2–20 min depending on the cell line).
-
Resuspend cells in fresh medium, pipette thoroughly to obtain single cell suspension. If your medium does not contain serum, you need to inactivate detaching agents, e.g., by addition of trypsin inhibitors. Measure total number of cells (see: counting cells).
-
Plate cells onto a new dish at the desired cell density.
Suspension cells
-
Collect cells with medium and briefly centrifuge (150–300 xg, 3–5 min) to pellet cells.
-
Remove medium and gently resuspend cells with a balanced salt solution. Briefly centrifuge (150–300 xg, 3–5 min) to pellet cells.
-
Remove salt solution and resuspend cells in fresh medium. Measure total number of cells (see: counting cells).
-
Plate cells onto a new dish at desired cell density.
It should be standard procedure to keep a record of the number of cell passages for all cell lines. For primary cell lines with a finite lifetime, it helps to monitor their viability and plan experiments before they reach senescence and cease dividing. For immortalized cells, it helps to monitor their age and design experiments up to a certain passage number.
Cell counting
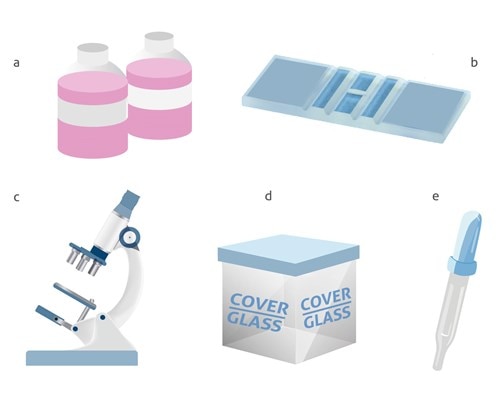
Despite the recent development of automated counters, the Neubauer chamber is still the most common method used for cell counting. A Neubauer chamber is used to perform a proper cell count, although the dimensions and volumes of each chamber may differ. The necessary elements to perform a cell count:
a) Cellular dilution to measure
b) Neubauer chamber
c) Optical microscope
d) Cover glass e) Pipette / micropipette with disposable tips
The equipment needed to perform a cell count with a Neubauer chamber
While performing the count, remember
Avoid counting cells that are obvious cell debris vs. rounded cells - When using trypan blue, count the cells that have excluded the dye
-
Cell suspensions should be dilute enough so that the cells do not overlap each other on the grid
-
Mix the cell suspension thoroughly before taking a sample to count
-
Count the cells in selected squares
-
Decide on a specific counting pattern to avoid bias
The formula for calculation of the cell concentration
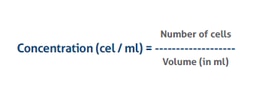
-
The number of cells is the sum of all cells counted in all squares -
-
The volume is the total volume of all the squares counted -
-
In case a dilution was applied, the final concentration needs to be converted to the original concentration before the dilution
The equipment needed to perform a cell count with a Neubauer
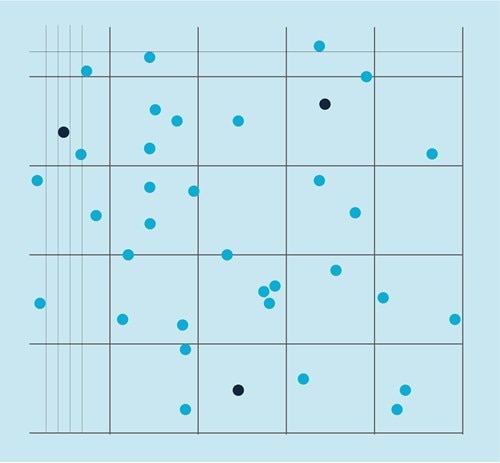
An example of cell countingwith a Neubauer chamber
-
We counted the cells in four squares to obtain a reliable estimate of the number of cells.
-
We obtained the following cell numbers: 54, 48, 52, and 60, which gave us a total of 214 cells.
-
The area of one square in the Neubauer chamber used equals 0.01 cm2, while the depth of the chamber is 0.01 cm.
-
Multiplying 0.01 cm2 by 0.01 cm gives 0.0001 ml total volume per one square.
-
Using the above equation, we divide the total number of cells (214) by volume (0.0001*4 squares = 0.0004) to obtain a concentration of 535 000 cells per ml.
Cell banks (cryopreservation)
Mammalian cell lines are suitable for long-term storage at low temperatures (below -130°C – usually in liquid nitrogen). Cell lines are routinely frozen to make and keep reference/parental cell lines, newly produced transgenic cell lines, keep stocks of primary and immortalized cells, and for shipping purposes. The viability of cell banks is dependent on the cryopreservation procedure employed when making them and on the proper storage conditions. It is good laboratory practice to create a number of master stocks at the lowest possible passage number for longterm storage together with a number of working stocks for general use in experiments.
Preparing cell banks
-
Grow cells toward the end of the logarithmic growth phase (~90% confluence). Ideally, use cells with the lowest possible passage number.
-
Passage cells as normal – detach adherent cells, collect and wash suspension cells (see: standard passaging).
-
Measure total cell number (see: counting cells).
-
Resuspend cells in freezing medium. In general, the freezing medium is routine culture medium supplemented with a cryoprotectant, an agent that prevents the formulation of ice crystals from the water present within cells that would destroy cell integrity. DMSO and glycerol are the two most commonly used cryoprotective agents. The exact concentration is cell-dependent and should be experimentally verified, but they are usually used at 5–10% (vol/vol) final concentration.
Note: Prepare freezing medium before adding to cells because concentrated DMSO is toxic to cells.
-
Aliquot cells into cryovials (usually 106–107 cells/ml) and gently cool down to -80°C. The most efficient cooling rate is 1°C per 1 minute, which can be achieved by putting vials into a commercial cryofreezing container or by wrapping them in a thick layer of cotton wool and placing in a -80°C freezer.
-
After 1–3 days place cells in a liquid nitrogen dewar and store in liquid nitrogen or the gas phase of liquid nitrogen for long-term storage.
Thawing cells from banks
-
Transport frozen cell vial in low temperature (portable liquid nitrogen container or dry ice) to cell culture area.
-
Prepare a culture dish with pre-warmed medium.
-
Thaw cells rapidly (e.g., in a 37°C water bath).
Note: Thawing cells rapidly ensures high cell viability.
-
Optional step to remove cryopreservant and non-viable cells: resuspend cells in medium and briefly centrifuge (150–300 xg for 3–5 min.). Resuspend cell pellet in fresh medium.
-
Transfer freshly thawed cells into a dish.
Cell transfection
Transfection of cells refers to the delivery of nucleic acid (DNA or RNA) into cultured cells. The most commonly used reagents are cationic lipids that can associate with nucleic acids to form positively charged complexes that allow interaction of DNA/RNA with the negatively charged cell membrane. That leads to the efficient entry of nucleic acids into cells via endocytosis.
Alternatively, nucleic acids can be delivered into cells by electroporation, co-precipitation of DNA with calcium phosphate, or polybrene/DMSO shock.
The delivered DNA does not usually integrate with the host genome. Therefore, DNA expression is generally transient. However, it is possible to engineer stable cell lines with transfected DNA stably integrated with the genome, but this requires a method of selecting cells with stable integration with a dedicated DNA vector design and antibiotic selection.