
COVID-19 Neuropathology Similar to Alzheimer’s Disease – The Role of the Ryanodine Receptor in “Long COVID”
Written by Leah Sittenfeld, M.D. Candidate at New York Medical College
As we draw near the third and hopefully final anniversary of the COVID-19 pandemic, there have been many reports of neurological symptoms and long-term sequelae in patients with SARS-CoV-2 infection. The neurologic manifestations of the disease range from mild olfactory and gustatory dysfunction1 to the more severe increased incidence of stroke2 and long-term cognitive dysfunction, often termed “long COVID.” There are likely many different mechanisms contributing to these neurological symptoms, including direct viral infection of neuronal tissue, indirect hypoxic-ischemic injury to the brain, and hyperactivation of the immune system leading to systemic inflammatory response syndrome2; interestingly, SARS-CoV-2 viral DNA is only detected in approximately 50% of patients’ brain tissue3, suggesting that direct viral infection is likely not a major mechanism of neuropathogenesis in COVID-19.
Ryanodine Receptor (RyR) Background – Setting the Stage
Recently, our laboratory conducted a small study examining the role of the ryanodine receptor (RyR) in COVID-19-associated cognitive dysfunction, uncovering a similar biochemical pattern to the pathology seen in Alzheimer’s disease. RyR is a homotetrameric intracellular calcium release channel on the sarco/endoplasmic reticulum that functions to exquisitely maintain the cytosolic calcium concentration in many tissues of the body, most notably skeletal muscle, cardiac muscle, and brain. Calcium is a key signaling molecule throughout the body, playing a critical role in muscle contraction in both the heart and skeletal muscles, as well as being important in neurotransmission and neuronal memory formation. Thus, dyshomeostasis of calcium regulation plays a major role in pathogenesis; and dysfunctional RyR has been implicated in many diseases, including heart failure4, muscular dystrophy5, and Alzheimer’s disease6. Post-translational modifications - primarily phosphorylation and oxidation - deplete the channel of the stabilizing subunit calstabin, resulting in a “leaky” phenotype that depletes the intracellular calcium stores and leads to cellular/organ dysfunction7. TGF-b, an anti-inflammatory cytokine involved in fibrosis and repair, activates the SMAD-dependent pathway and upregulates reactive oxygen species (ROS)8. This increase in ROS leads to the pathological oxidation of RyR which has been shown to contribute to disease states in previous research9.
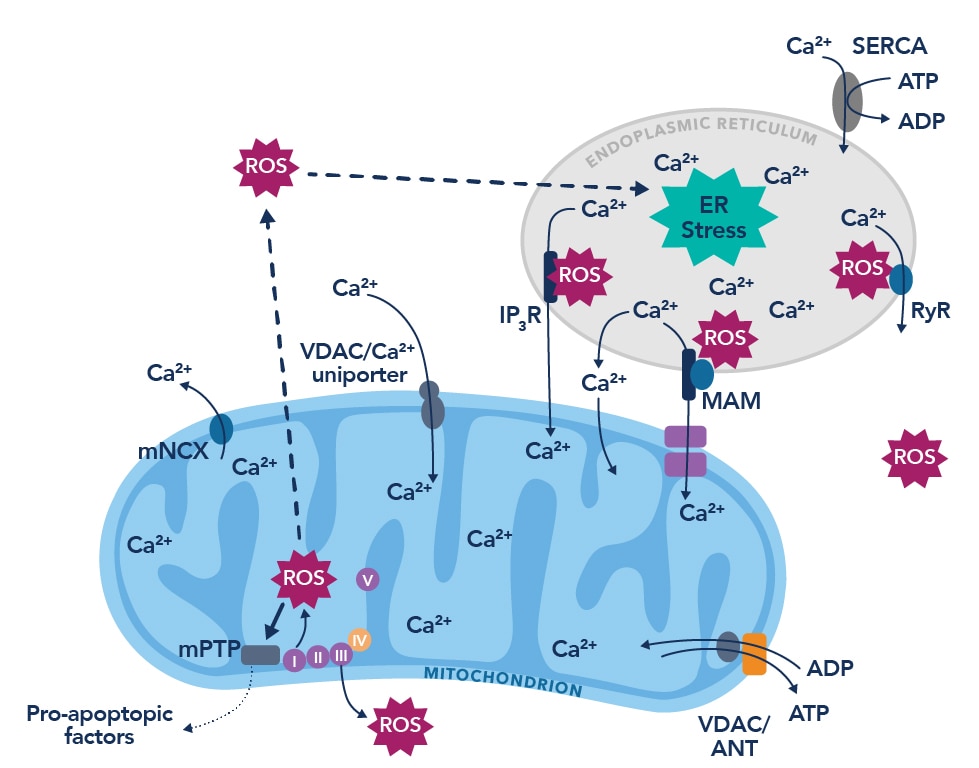
Figure 1. The vicious cycle of endoplasmic reticulum calcium leak and the production of reactive oxygen species. Figure adapted from Görlach et al., 201510.
Common Pathway Activation Observed in Alzheimer’s Disease and COVID-19
SARS-CoV-2 uses the ACE2 receptor to enter the body and exploits the host’s replicative capacity to spread its infection. In COVID-19 patients, ACE2 receptor downregulation was seen with concurrent TGF-b upregulation, with those patients with severe symptoms having higher serum TGF-b levels11,12. In addition, reduced ACE2 has been associated with hyperphosphorylated tau and increased amyloid-beta (Ab)13, the neuropathological hallmarks of Alzheimer’s disease. Hyperphosphorylated tau is characteristically “sticky” and leads to the formation of intracellular neurofibrillary tau tangles, disrupting cellular function and ultimately leading to neuronal cell death. Amyloid-beta accumulates extracellularly between neurons, and while its mechanism is not completely understood, it has been shown to activate inflammatory pathways in the brain14, again leading to neuronal cell death. Taken together, reduced ACE2, increased TGF-b with subsequent oxidation of RyR, and a prior link between leaky RyR and Alzheimer’s disease warranted further investigation into the biochemical pathways leading to the neuropathology observed in patients who succumbed to COVID-19 infection with no previous history of neurodegenerative disease.
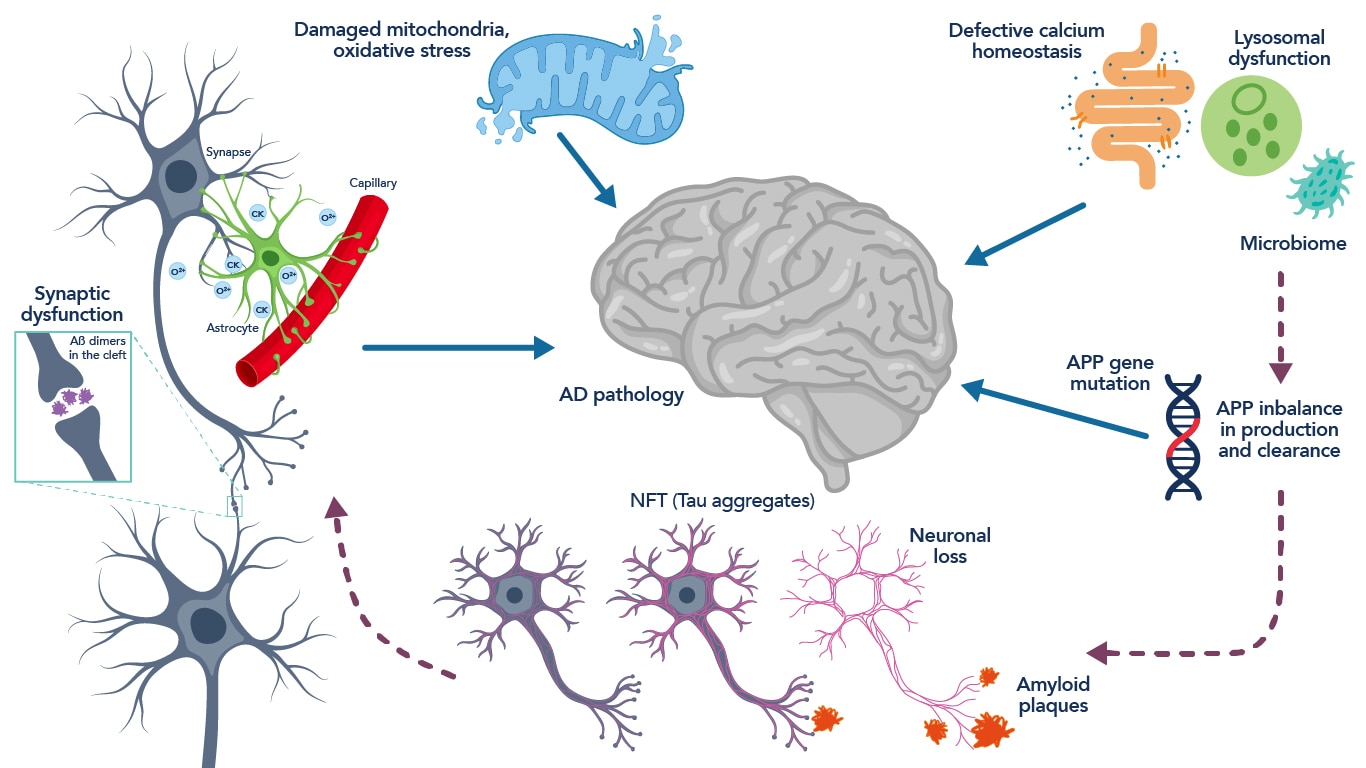
Figure 2. Schematic representation of the neuropathology and pathophysiology associated with Alzheimer’s disease. Figure adapted from Hany et al., 201619.
Biochemical Findings Implicating RyR in COVID-19 Neuropathophysiology
We analyzed samples from the cortex and cerebellum of deceased COVID-19 patients using immunoblotting techniques and demonstrated decreased calstabin binding and increased phosphorylation, oxidation, and nitrosylation of RyR2, as well as increased activation of the ROS-generating SMAD pathway, as evidenced by elevated levels of phosphorylated SMAD, compared to controls. Furthermore, analyses of these samples also revealed significantly increased levels of hyperphosphorylated tau in both the cortex and cerebellum, a finding unique from Alzheimer’s disease which classically only demonstrates hyperphosphorylated tau in cortex15. Ex vivo treatment of the samples with ARM210, a novel small molecule Rycal drug developed by our laboratory, restored calstabin binding to the posttranslationally modified channels but had no effect on the other modifications. This drug has previously been shown to restore channel function from a “leaky” phenotype16 and represents a potential therapeutic approach for addressing cognitive dysfunction in COVID-19.
Continuing Research and Future Directions
Recent studies outside of our laboratory have also found evidence of increased incidence of Alzheimer’s disease in COVID-19 patients, particularly in elderly patients17. Furthermore, there have been reports of increased inflammasome-mediated inflammation that leads to imbalances in kinases and phosphatases that contribute to the formation of neurofibrillary tangles akin to those seen in Alzheimer’s disease18. Our research proposes a tangible pathway involving a vicious cycle of RyR2 dysfunction, oxidative stress, and activation of hyperphosphorylation pathways associated with Alzheimer’s disease pathology. This mechanism of injury is consistent with previous research our laboratory has conducted on Alzheimer’s disease6 and represents a unique understanding of the pathology underlying the cognitive dysfunction exhibited by many COVID patients. Using these findings, future work can be directed towards targeting therapeutic intervention to this pathway to alleviate the dysfunction and prevent the initiation or furthering of neurodegenerative sequelae.
Antibodies to study RyR
|
|
CoraLite® Plus 647 RYR2 Polyclonal Antibody
|
|
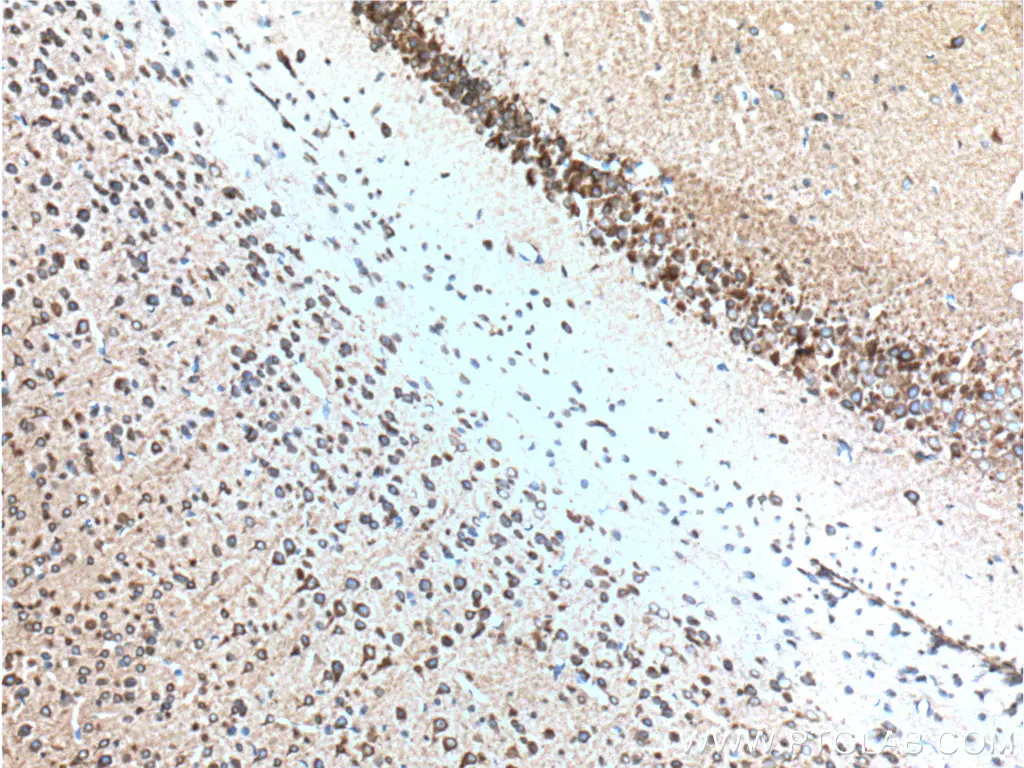
IHC staining of paraffin-embedded mouse brain tissue using RYR1 antibody (26968-1-AP) |
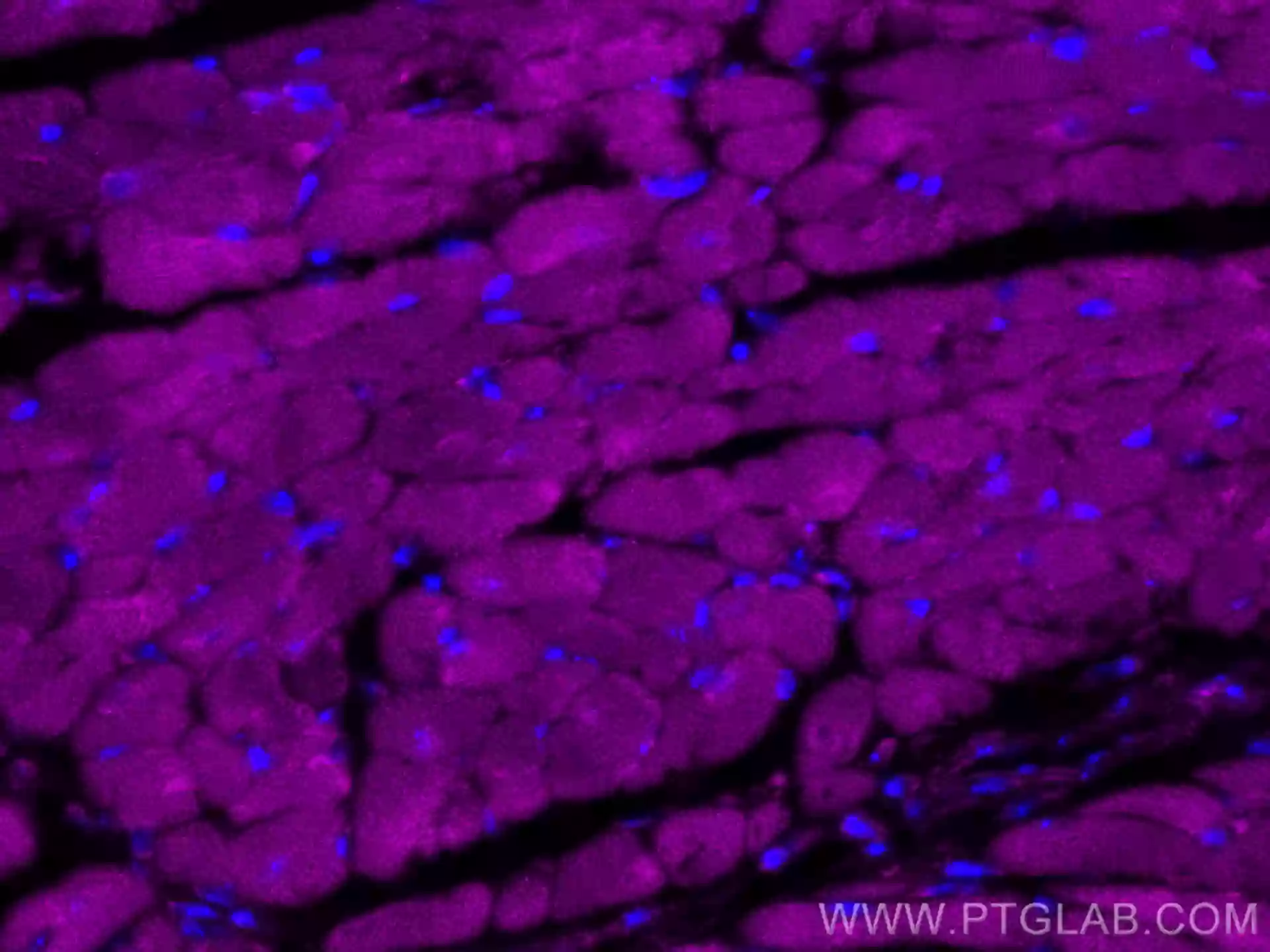
IF of fixed mouse heart tissue using CoraLite® Plus 647 RYR2 antibody (CL647-27587) |
References
1. Lou, J. J. et al. Neuropathology of COVID-19 (neuro-COVID): clinicopathological update. Free Neuropathol 2 (2021). https://doi.org:10.17879/freeneuropathology-2021-2993
2. Wu, Y. et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun 87, 18-22 (2020). https://doi.org:10.1016/j.bbi.2020.03.031
3. Matschke, J. et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol 19, 919-929 (2020). https://doi.org:10.1016/s1474-4422(20)30308-2
4. Dridi, H. et al. Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat Rev Cardiol 17, 732-747 (2020). https://doi.org:10.1038/s41569-020-0394-8
5. Andersson, D. C. et al. Leaky ryanodine receptors in β-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skelet Muscle 2, 9 (2012). https://doi.org:10.1186/2044-5040-2-9
Related Content
Neuropilin-1 joins ACE2 as SARS-CoV-2 gateway
How Nanobodies could advance Sars-CoV diagnostics
Replication Race: Scientists unravel why Omicron spreads faster
The multi-organ impact of COVID-19
Support
Newsletter Signup
Stay up-to-date with our latest news and events. New to Proteintech? Get 10% off your first order when you sign up.