
Vascular dysfunction in Alzheimer’s Disease
Written by Katy Walsh, PhD student researching vascular dementia at the University of Manchester, UK.
Alzheimer’s Disease (AD) is the leading cause of dementia, affecting over half a million people in the UK alone 1. Despite the huge toll of this devastating disease on the individuals affected, their families, and healthcare providers, there is still no cure or treatment for its development. As AD is primarily a disease of the aged, both the numbers and cost of care are expected to grow rapidly as the aging population continues to increase. It is therefore vital that researchers continue to investigate the mechanisms underlying this disease to identify novel therapeutic targets.
What is Alzheimer’s Disease?
AD is characterized by an accumulation of the amyloid beta (Ab) peptide within the neuronal space. Ab is produced in everyone by cleavage of the amyloid precursor protein (APP) by two enzymes: b secretase and g secretase. The second cleavage by g secretase is less specific, leading to the production of different-length isoforms of Ab. The most abundant is Ab42, which aggregates in plaques on the neurons in the brains of patients with AD (Figure 1). These plaques are thought to interfere with the neurons’ ability to communicate signals, leading to neuronal cell death and eventual cognitive decline. However, treatments designed to prevent the production of Ab42 or improve its clearance from the brain have proved unsuccessful at preventing or slowing disease progression, suggesting other mechanisms are involved in the decline of brain function.
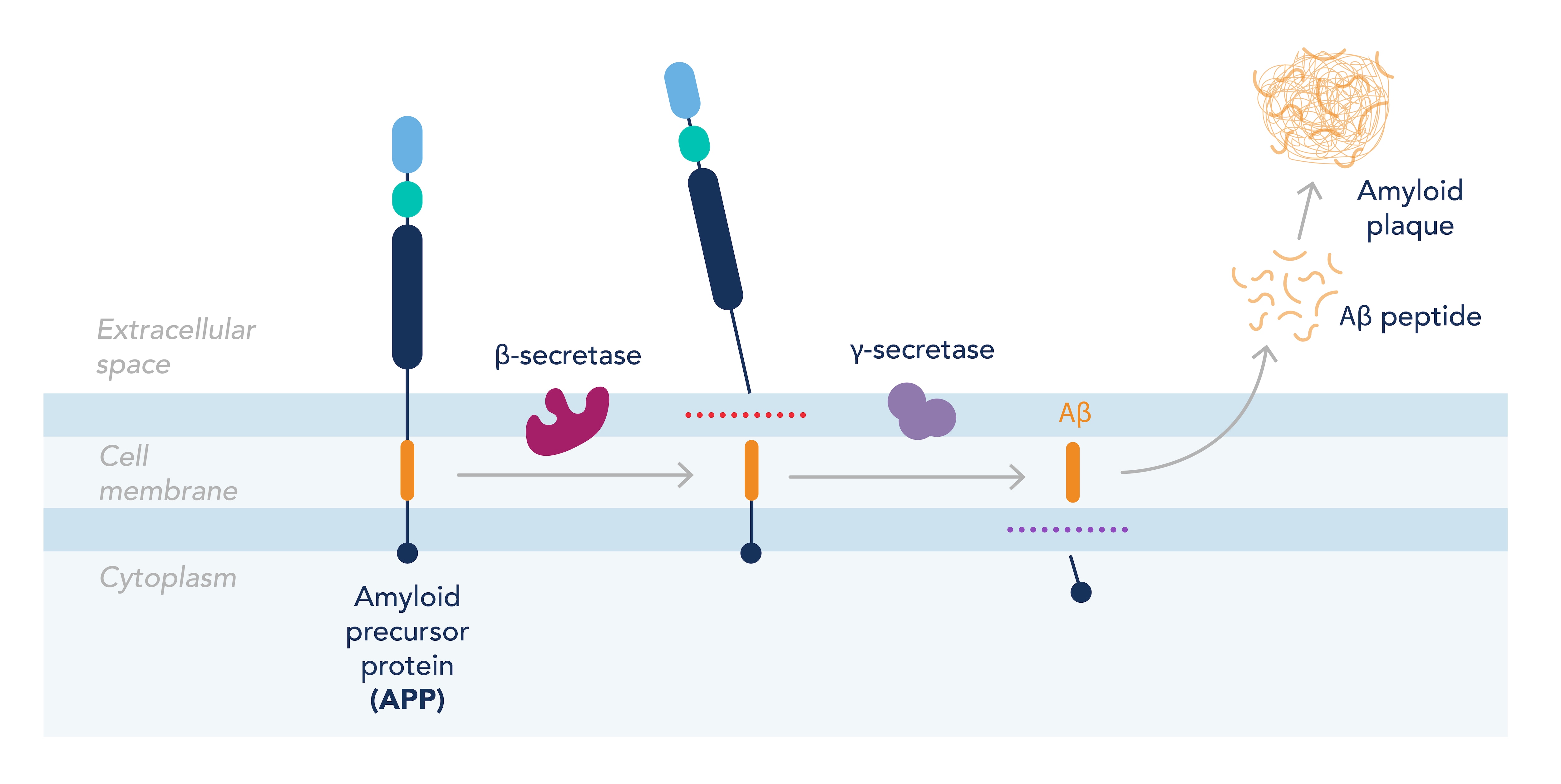
Figure 1. Overview of the formation of amyloid plaques from the dysregulation of APP processing.
Cerebral blood flow is tightly regulated
Interestingly, one of the earliest detectable signs of AD is a reduction in cerebral blood flow (CBF). Unlike other organs, the brain cannot store energy and therefore requires a constant and adaptable supply of blood to deliver the oxygen and glucose required to meet its demands. This means CBF must be strictly regulated to rapidly respond to different stimuli. The arteries within the brain do this by two specific mechanisms: autoregulation and neurovascular coupling (NVC). Autoregulation refers to arterial vasoconstriction or vasodilation at different physiological pressures to maintain CBF. Arteries can do this due to myogenic tone – a partially constricted state allowing further constriction or dilation in response to different stimuli. As pressure increases, i.e., when stressed, the cerebral arteries constrict to maintain constant flow and protect the brain. NVC is the cerebral arteries’ ability to detect local changes in brain activity and vasodilate to increase blood flow to that area to meet the metabolic demands of the surrounding neurons. Unfortunately, both autoregulation and NVC are disrupted in AD patients, supporting the idea that vascular dysfunction underlies disease progression.
Vascular dysfunction in Alzheimer’s Disease
There are multiple reasons to believe that vascular dysfunction could be responsible for cognitive decline in AD patients. Firstly, the majority of risk factors for cardiovascular disease are also risk factors for AD, including obesity, smoking, hypertension, and diabetes, suggesting a link between the two pathologies 2. Interestingly, lowering blood pressure by anti-hypertensive treatment has been shown to decrease the development of cognitive impairment 3. Additionally, vascular lesions such as white matter hyperintensities and microbleeds correlate positively with disease progression and are a more accurate predictor of disease severity compared with the diagnostic values of brain volume and atrophy 4. Finally, increased capillary stalling was shown in an animal model of AD, where capillaries are temporarily blocked due to the adhesion of immune cells to the vascular wall 5. This stalling was reduced when the mice were treated with an antibody against the immune cell, resulting in increased CBF and cognitive function.
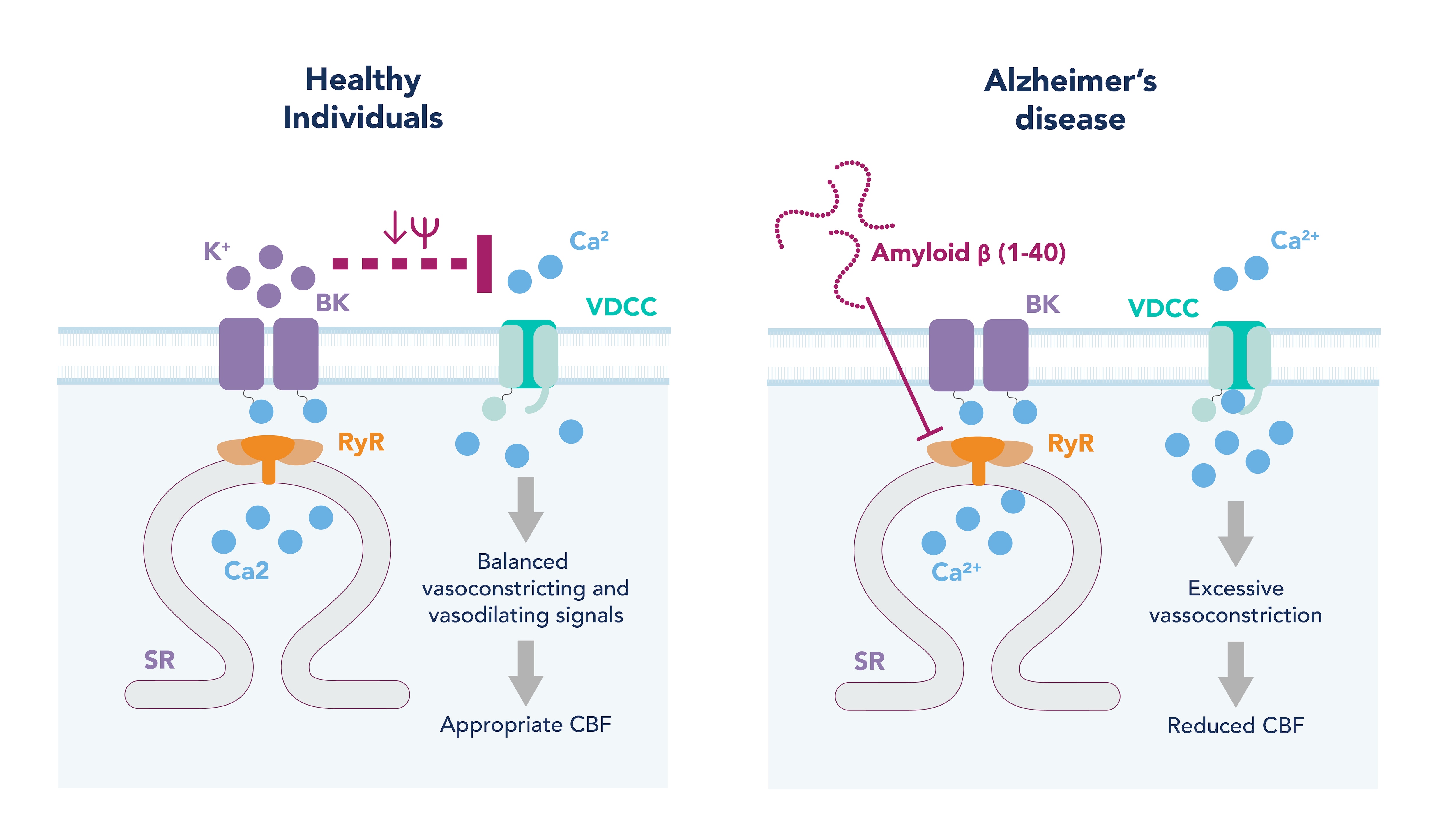
Figure 2. Potential mechanism of cerebral vascular dysfunction in Alzheimer’s disease.
How is vascular function disrupted in Alzheimer’s Disease?
While Ab42 is the most abundant, Ab40 is the second most produced isoform and is described as more soluble and associated with the cerebral vasculature. Cerebral amyloid angiopathy (CAA) is generally thought of as a separate disease from AD, characterized by a build-up of Ab40 in the cerebral vascular walls. However, CAA has been found in 80-100% of AD patient brains 6, and Ab40 levels correlate better with CBF reduction in AD compared to Ab42 7,8. This suggests CAA and AD could exist as a single pathology and Ab40 could be a driver of disease progression.
In healthy arteries, when intraluminal pressure is increased, this is sensed by the vascular smooth muscle cells (VSMC), leading to their depolarization. This depolarization opens the voltage-dependent calcium channels (VDCC) on the plasma membrane, leading to calcium influx. An increase in intracellular calcium in VSMC activates the contractile machinery in the cell causing contraction and vasoconstriction. To prevent excessive constriction in response to this pressure, a negative feedback loop exists whereby the increase in intracellular calcium also activates and opens ryanodine receptors on the sarcoplasmic reticulum (SR). This allows for a calcium efflux from SR into the cytoplasm known as a calcium spark. Calcium sparks can activate large-conductance calcium-activated potassium (BK) channels on the plasma membrane, causing a potassium efflux from the cell. This potassium efflux hyperpolarizes the cell and closes the VDCC, causing the VSMC to relax (Figure 2, left hand image).
In a mouse model of AD that overexpresses APP, both myogenic tone (and therefore autoregulation) and NVC are disrupted. Myogenic tone is increased in these diseased mice, and is shown to be due to a decrease in the calcium spark frequency and therefore compensatory negative feedback loop signals, leading to enhanced VSMC contraction and vasoconstriction 9 (Figure 2, right hand image). Interestingly, this disruption in calcium signaling is observed in the arteries of healthy mice when exposed to Ab40 for just 30 minutes. Furthermore, Ab40, but not Ab42, constricts cerebral arteries and decreases CBF when placed on the surface of the brain of healthy mice 10. This suggests Ab40 is responsible for the vascular dysfunction and reduction in CBF observed in AD patients, and therapeutics that target the vasculature and Ab40’s action may prove more fruitful than previous Ab42-targeting compounds.
Related Products
|
Alzheimer’s disease |
Arterial sensing |
Neurovascular coupling |
References
- BHF. British Heart Foundation UK Factsheet 2020. Br. Hear. Found. (2020).
- Pase, M. P., Satizabal, C. L. & Seshadri, S. Role of Improved Vascular Health in the Declining Incidence of Dementia. Stroke (2017) doi:10.1161/STROKEAHA.117.013369.
- Williamson, J. D. et al. Effect of Intensive vs Standard Blood Pressure Control on Probable Dementia. JAMA 321, 553 (2019).
- Brickman, A. M. et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch. Neurol. (2012) doi:10.1001/archneurol.2012.1527.
- Cruz Hernández, J. C. et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. (2019) doi:10.1038/s41593-018-0329-4.
- Jellinger, K. A. Alzheimer disease and cerebrovascular pathology: An update. Journal of Neural Transmission (2002) doi:10.1007/s007020200068.
- Niwa, K. et al. Aβ1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. U. S. A. (2000) doi:10.1073/pnas.97.17.9735.
- Alonzo, N. C., Hyman, B. T., Rebeck, G. W. & Greenberg, S. M. Progression of cerebral amyloid angiopathy: Accumulation of amyloid- β40 in affected vessels. J. Neuropathol. Exp. Neurol. (1998) doi:10.1097/00005072-199804000-00008.
- Taylor, J. L. et al. Functionally linked potassium channel activity in cerebral endothelial and smooth muscle cells is compromised in Alzheimer’s disease. Proc. Natl. Acad. Sci. 119, (2022).
- Niwa, K. et al. Aβ-peptides enhance vasoconstriction in cerebral circulation. Am. J. Physiol. Circ. Physiol. 281, H2417–H2424 (2001).
Related Content
COVID-19 Neuropathology Similar to Alzheimer’s Disease
How to Identify Activated Microglia
7 Tips to successfully culture primary rodent neurons
Support
Newsletter Signup
Stay up-to-date with our latest news and events. New to Proteintech? Get 10% off your first order when you sign up.
Able™