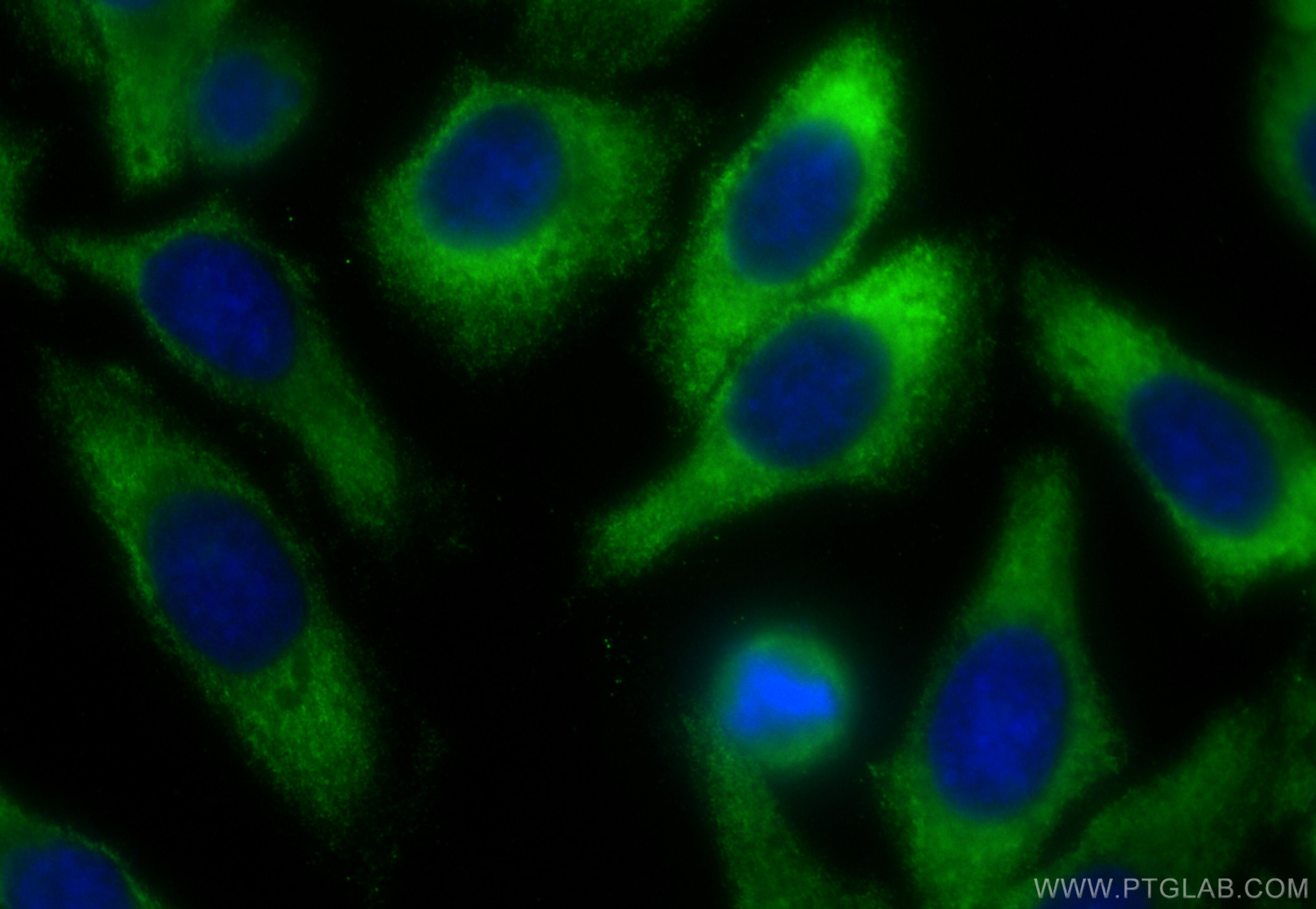
Tested Applications
| Positive IF/ICC detected in | HepG2 cells |
Recommended dilution
| Application | Dilution |
|---|---|
| Immunofluorescence (IF)/ICC | IF/ICC : 1:50-1:500 |
| It is recommended that this reagent should be titrated in each testing system to obtain optimal results. | |
| Sample-dependent, Check data in validation data gallery. | |
Product Information
CL488-66121 targets Alpha Galactosidase A in IF/ICC applications and shows reactivity with human samples.
| Tested Reactivity | human |
| Host / Isotype | Mouse / IgG2a |
| Class | Monoclonal |
| Type | Antibody |
| Immunogen |
CatNo: Ag7505 Product name: Recombinant human GLA protein Source: e coli.-derived, PET28a Tag: 6*His Domain: 81-425 aa of BC002689 Sequence: WKDAGYEYLCIDDCWMAPQRDSEGRLQADPQRFPHGIRQLANYVHSKGLKLGIYADVGNKTCAGFPGSFGYYDIDAQTFADWGVDLLKFDGCYCDSLENLADGYKHMSLALNRTGRSIVYSCEWPLYMWPFQKPNYTEIRQYCNHWRNFADIDDSWKSIKSILDWTSFNQERIVDVAGPGGWNDPDMLVIGNFGLSWNQQVTQMALWAIMAAPLFMSNDLRHISPQAKALLQDKDVIAINQDPLGKQGYQLRQGDNFEVWERPLSGLAWAVAMINRQEIGGPRSYTIAVASLGKGVACNPACFITQLLPVKRKLGFYEWTSRLRSHINPTGTVLLQLENTMQMSL Predict reactive species |
| Full Name | galactosidase, alpha |
| Calculated Molecular Weight | 49 kDa |
| Observed Molecular Weight | 49 kDa |
| GenBank Accession Number | BC002689 |
| Gene Symbol | GLA |
| Gene ID (NCBI) | 2717 |
| RRID | AB_2883263 |
| Conjugate | CoraLite® Plus 488 Fluorescent Dye |
| Excitation/Emission Maxima Wavelengths | 493 nm / 522 nm |
| Excitation Laser | Blue laser (488 nm) |
| Form | Liquid |
| Purification Method | Protein A purification |
| UNIPROT ID | P06280 |
| Storage Buffer | PBS with 50% glycerol, 0.05% Proclin300, 0.5% BSA, pH 7.3. |
| Storage Conditions | Store at -20°C. Avoid exposure to light. Stable for one year after shipment. Aliquoting is unnecessary for -20oC storage. |
Background Information
GLA, also named as Melibiase and Alpha-galactosidase A, belongs to the glycosyl hydrolase 27 family. It hydrolyzes terminal, non-reducing alpha-D-galactose residues in alpha-D-galactosides, including galactose oligosaccharides, galactomannans and galactolipids. Fabry disease is an X-linked lysosomal storage disorder resulting from the deficient activity of GLA. Enzyme replacement therapy (ERT) with GLA is currently the most effective therapeutic strategy for patients with Fabry disease, a lysosomal storage disease.
Protocols
| Product Specific Protocols | |
|---|---|
| IF protocol for CL Plus 488 Alpha Galactosidase A antibody CL488-66121 | Download protocol |
| Standard Protocols | |
|---|---|
| Click here to view our Standard Protocols |