Home / Video Hub
Videos

Prof. Richard Lea: The Future of Fertility
In this episode, we sit down with Professor Richard Lea, leading reproductive biologist and star of Channel 4’s Celebrity Save Our Sperm, to explore the urgent and often overlooked crisis in male reproductive health. With sperm counts dropping globally and our environmental impact mounting, Professor Lea brings both scientific expertise and real-world insight to the conversation. Whether you're a researcher, future parent, or just curious about what’s happening below the belt, this episode is packed with eye-opening facts, surprising stories, and a clear-eyed look at the future of fertility.
Western BlotView all






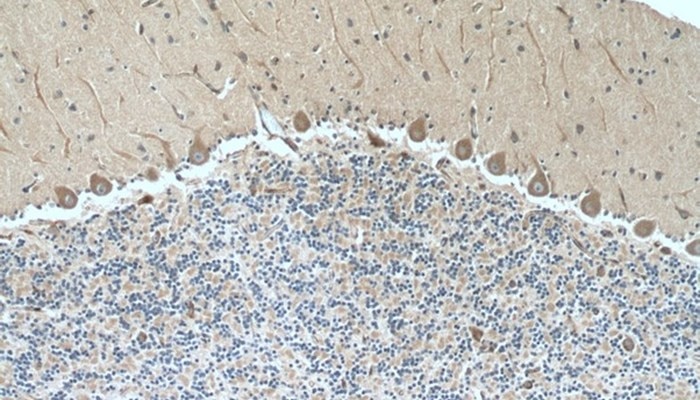
ImmunohistochemistryView all
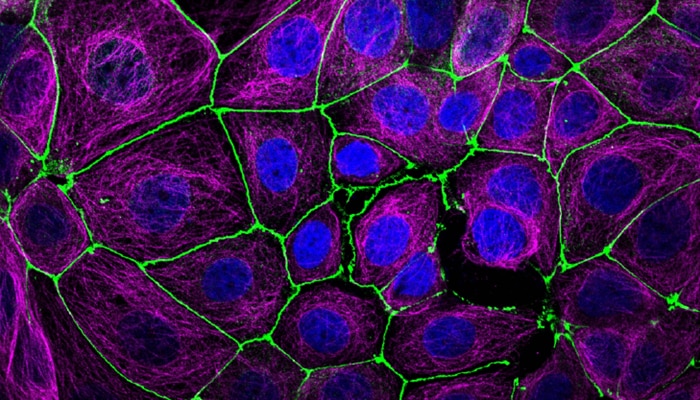

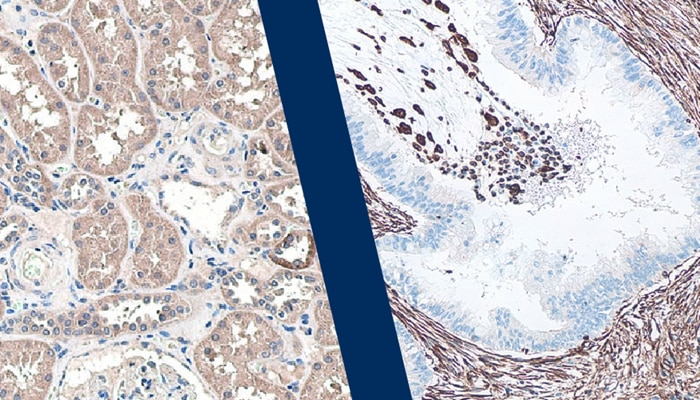






ImmunoprecipitationView all
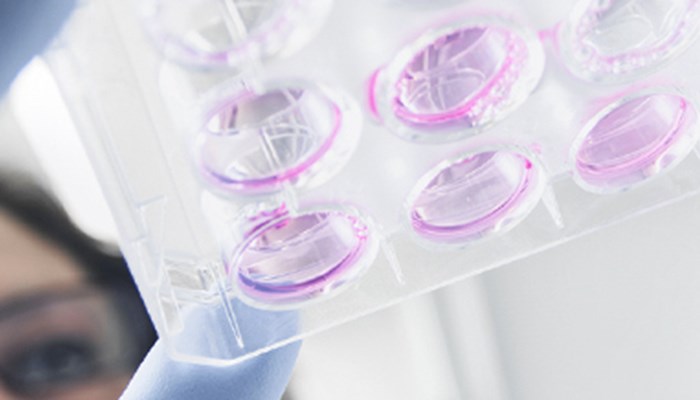





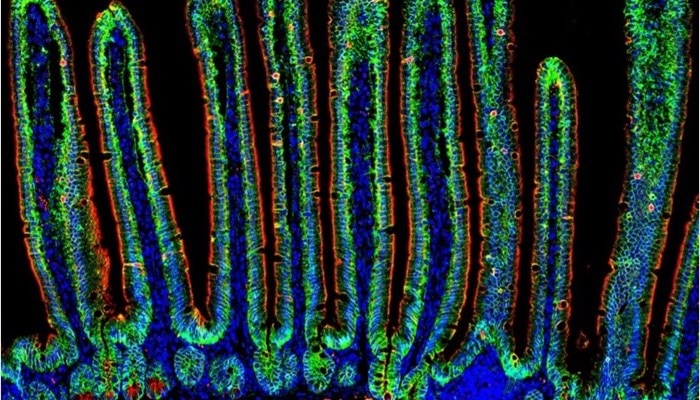
ImmunofluorescenceView all
 59:29
59:29
Meet the Expert: Dr. Indra Chandrasekar - Exploring the kidney cellular and organelle architecture using immunofluorescence imaging techniques
Technical Talk



OtherView all