
How to solve the genome DNA damage
DNA damage repair mechanisms: in the prevention of age-related diseases and tumorigenesis
DNA damage repair mechanisms:
Click the links to navigate:
Browse more DNA damage repair markers here |
Unrepaired DNA damage leads to mutagenesis or cell death. DNA damage accumulation in myocytes or neurons can result in degenerative diseases (1). Every day, cells are exposed to a broad range of DNA-damaging factors (2). What are the main mechanisms of DNA repair? (Figure 1).
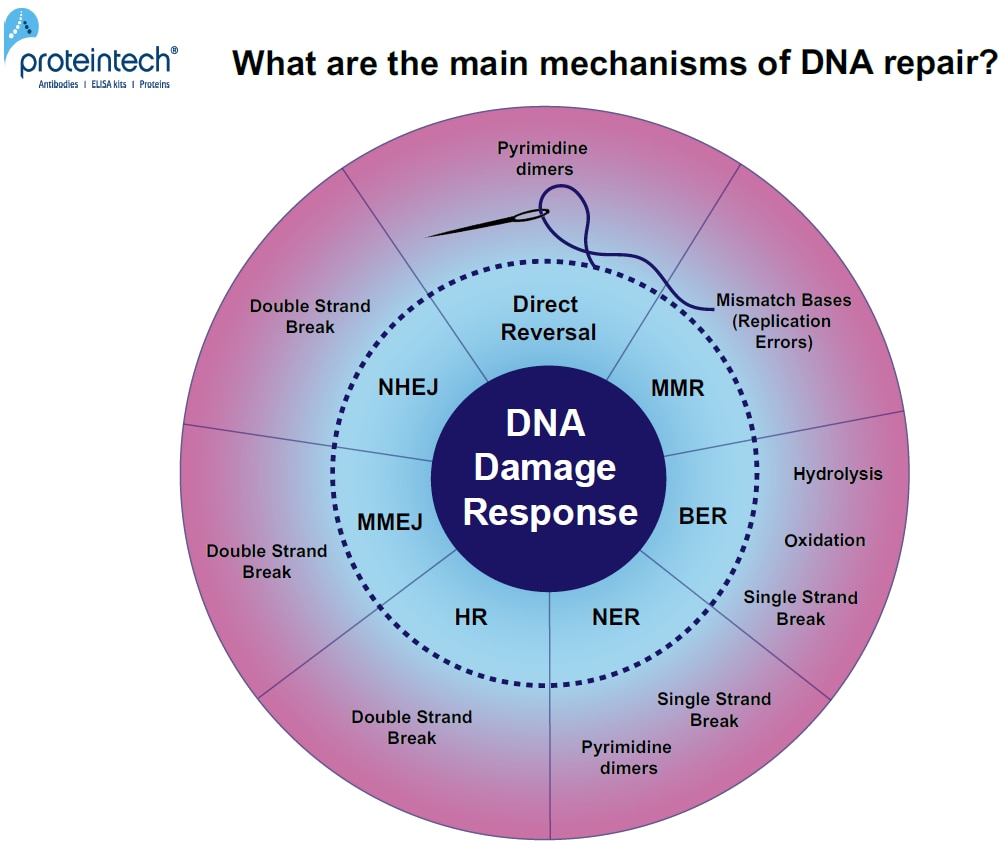
FIGURE 1. Overview of DNA damage-specific repair pathways. Response mechanisms: Direct reversal, MMR (MisMatch Repair), BER (Base Excision Repair), NER (Nucleotide Excision Repair), HR (Homologous Recombination), MMEJ (Microhomology-Mediated End Joining), and NHEJ (Non-Homologous End Joining).
DNA damage repair mechanisms
The cell recognizes and marks DNA damage sites, attracting key players in DNA damage response. The activation of cell cycle checkpoints and cell cycle arrest extends the time available to the repair machinery (Figure 2). DNA repair can occur at several stages of the cell cycle but cells are mostly arrested in the G1/S and G2/M phases. If damage occurs due to replication errors, an arrest can also occur in the S-phase or later in mitosis.
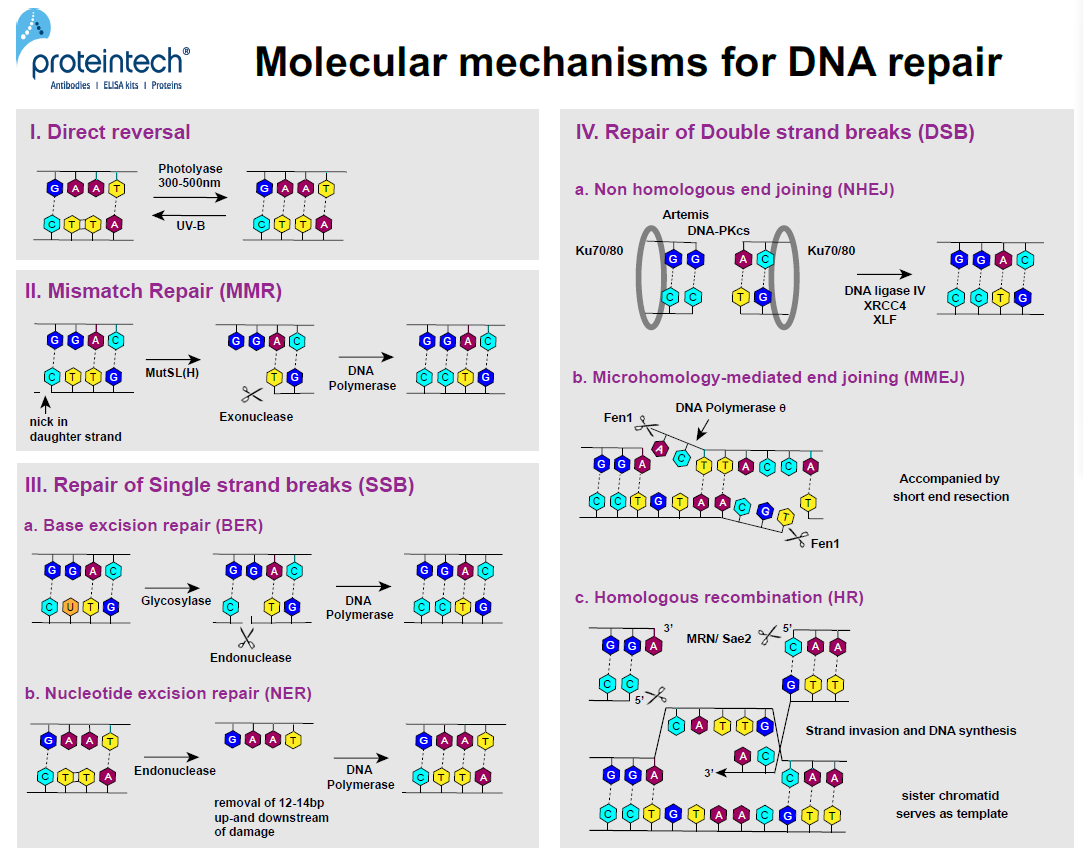
FIGURE 2. DNA repair mechanisms at the molecular level.
I. Direct reversal
Nobel prize winner Aziz Sancar was the first to discover the E. coli enzyme photolyase (3). In response to UV exposure, two adjacent pyrimidine residues can form covalent bonds resulting in pyrimidine dimers (Py-Py). Photolyase is activated by 300–500nm wavelengths. The enzyme has the ability to cleave covalent bonds of Py-Py dimers within subnanoseconds without breaking the phosphodiester bond (4). Today we know that direct reversal is a repair mechanism in both prokaryotes and eukaryotes, although not in humans as NER (nucleotide excision repair) is used instead (Figure 2; I).
II. Mismatch Repair (MMR)
MMR was one of the major discoveries by Paul Modrich (Nobel prize 2015). It is based on the cell’s ability to discriminate between the parental DNA strand and the newly synthesized “daughter” strand during the replication process. In bacteria, methylation features only in the parental strand and serves as a marker for proofreading systems to identify the DNA template and search for mismatches in the “daughter” strand. There is no evidence of methylation in eukaryotes; breaks that occur during replication in both lagging and leading strands play a role in the recruitment of proofreading systems. The orchestrated action of two dimers, MutS and MutL (also MutH in E. coli), leads to the recognition of mismatch bases in the “daughter” strand and creates an incision at specific sites up to 1kb from the mismatch base. Exonucleases remove the part of the DNA from the incision to the mismatch, allowing DNA polymerase d (Polymerase III in prokaryotes) to finally re-synthesize the missing part (Figure 2; II).
III. Repair of Single Strand Breaks (SSB)
If a single strand break occurs, the side of the damage is bound and marked first by ATR (Ataxia Telangiectasia and Rad3-related) and RPA (Replication Protein A). ATR becomes active upon autophosphorylation and forms a dimer with ATRIP(ATR Interacting Protein), subsequently recruiting the 9-1-1 complex (composed of Rad9, Hus1, and Rad1) to the SSB side (5). ATR phosphorylates Chk1 (Checkpoint Kinase 1), which eventually leads to cell cycle arrest through the inhibition of Cdc25 (6). SSBs can subsequently be resolved via the two alternative methods outlined below:
a) Base Excision Repair (BER)
BER, first described by Tomas Lindahl, mainly occurs in response to single nucleotide damage (7). Glycosylases travel along and flip the bases of each DNA strand in order to evaluate the status of each base pair. If a faulty base is detected, it is removed, and an AP endonuclease cuts the phosphodiester backbone. DNA polymerase will subsequently fill in the correct nucleotide. This repair mechanism can also be used to fix depurination (a missing purine base), thus eliminating the need for glycosylases (Figure 2; IIIa).
b) Nucleotide Excision Repair (NER)
The heterodimer DDB (DNA Damage-Binding) is implicated in the recognition of bulkier DNA damage (e.g., pyrimidine dimerization). It is involved in the ubiquitination of the core histones H2A, H3, and H4, as well as XPC. This results in NER activation, starting with the removal of 12-24 nucleotides around the damaged DNA region. Subsequently, RFC (Replication Factor C) and PCNA (Proliferating Cell Nuclear Antigen) bind to the DNA, which enables the gap to be refilled by DNA polymerase (d, e, or k) (Figure 2; IIIb).
IV. Repair of Double Strand Breaks (DSB)
ATM (Ataxia-Telangiectasia-Mutated) is the key player in DNA damage response due to DSB. The inactive form of ATM usually exists as a multimer, which dissociates upon autophosphorylation and activation during DNA damage (5). ATM is next recruited to DSB sites where it phosphorylates the H2AX histone variant. The phosphorylation of H2AX to gH2AX is a quick response and thus serves as an excellent DNA damage marker. ATM also activates Chk2 (Checkpoint Kinase 2) and/or p53 via phosphorylation. This results in Cdc25 inhibition, leads to cell cycle arrest, and subsequently to DNA damage repair or apoptosis. Depending upon when in the cell cycle the DSB occurs, one of the following pathways is used for its repair:
a) Non-Homologous End Joining (NHEJ)
The Ku-heterodimer (Ku70-Ku80) binds to the sides of the DSB, forming a ring-shaped hub for NHEJ factors (8). Ku binds to DNA regardless of the sequence and can occur at blunt ends, as well as at 5’ or 3’ overhangs. It recruits DNA-PKcs, which is another PI3K-related kinase. After recruitment and phosphorylation of the nuclease Artemis, single strand overhangs are removed and the two ends are bonded by Ligase IV, XRCC4, and XLF. NHEJ is a non-preservative DNA repair pathway as it usually results in the loss of the damaged area (Figure 2; IVa).
b) Microhomology-Mediated End Joining (MMEJ)
During MMEJ, regions of microhomology are identified on one side of the DSB and single-stranded overhangs are aligned accordingly. DNA polymerase q helps to anneal and ligate the aligned regions of microhomology.The nucleaseFen1(Flap Endonuclease 1) removes the single-stranded DNA flaps, enabling DNA polymerase q to fill the gaps. This repair pathway is always accompanied by short end resection, which leads to deletions within the DNA strand, causing a mutation (Figure 2; IVb).
c) Homologous Recombination (HR)
HR usually occurs in G2. It is less prone to errors than the two other DSB repair pathways. It uses the sequence of the sister chromatid as a template. First, the MRN complex (Mre11, Rad50, and Nbs1) binds to one side of the break. MRN and Sae2 next create short 3’ overhangs by chopping the 5’ ends around the DSB. RPA and Rad51 coat the single strands and form a nucleoprotein filament. This filament invades the intact double strand DNA of the “sister” chromatid upon finding the homologous region. Subsequently, RFC, PCNA, and DNA polymerases (d, e, or a) bind to the side of the strand invasion and synthesize the missing regions using the strand of the intact “sister” chromatid as a “template” (Figure 2; IVc).
What if the DNA damage repair mechanisms fail?
An accumulation of DNA damage is commonly related to cancer or degenerative diseases and usually affects a specific cell type or organ. In contrast, the non-functionality of one or more DNA repair proteins, as a result of a genetic disorder, will impair DNA damage repair in all types of cells simultaneously. As an example, genetic diseases like Xeroderma pigmentosum, Cockayne syndrome, or Trichothiodystrophy exhibit defects in the Nucleotide Excision Pathway (NER) (9, 10). NER is generally involved in resolving single strand breaks and pyrimidine dimers, which are primarily inflicted by UV-B. Such patients are highly photosensitive and prone to developing skin cancer.
Fanconi Anemia is another example of a genetic disease where several proteins involved in Homologous Recombination (HR) can be affected. Symptoms include developmental abnormalities and bone marrow failure.
Even more severe are ATM and ATR mutations causing Ataxia Telangiectasia and Seckel syndrome. They are related to growth defects, neurodegeneration, microcephaly, and high cancer risk. There are still many questions regarding DNA damage and the repair process that will be crucial in finding future cures for DNA damage mutations and abnormalities (Table 1).
References
1. Stein and Toiber (2017) “DNA damage and neurodegeneration: the unusual suspect”.
2. Lindahl and Barnes (2000) “Repair of endogenous DNA damage“.
4. Kao et al. (2005) “Direct observation of thymine dimer repair in DNA by photolyase”
5. Qiu et al. (2018) “ATR/CHK1 inhibitors and cancer therapy”.
6. Awasthi et al. (2015) “ATM and ATR signaling at a glance”.
7. Lindahl (2012) “My Journey to DNA Repair”
8. Frit et al (2019) “Plugged into the Ku-DNA hub: The NHEJ network”
9. Menck and Munford (2014) “DNA repair diseases: What do they tell us about cancer and aging?”
10. Torgovnick and Schumacher (2015) “DNA repair mechanisms in cancer development and therapy”
Related Content
CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing
Epigenetics Modifications - DNA methylation
Epigenetics - DNA layers and chromatin modeling
Using Flow Cytometry with Epigenetics

Support
Newsletter Signup
Stay up-to-date with our latest news and events. New to Proteintech? Get 10% off your first order when you sign up.
Able™